What is "phenylketonuria"?
Phenylketonuria is a congenital genetic disease that is associated with a disorder of amino acid metabolism. The substance enters the human body as part of protein foods. As a result, phenylalanine and its toxic derivatives accumulate. As a result, damage to the central nervous system is observed. The pathology is characterized by mental retardation, cognitive and behavioral disorders. Children have a clear lack of desire and thought to compare everything around and analyze. Passive interest may appear, but nothing more. There is a problem with perception and adaptation to society. Gradually, the emotional sphere also suffers. There is instability in behavior.
In most cases, the classic form of the disease develops. With it, a decrease in liver enzyme activity is observed. It is called phenylanine-4-hydroxylase.
There are other types of pathology. In 1% of cases, an atypical type of the disease develops. It appears as a result of a mutation of another type of gene. It is responsible for the enzyme coding process. The disease is inherited according to an autosomal recessive pattern.
Due to the presence of a metabolic block, side pathways of phenylalanine metabolism are activated. This leads to the accumulation of toxic derivatives of its action. In particular, they are phenyllactic and phenylpyruvic acids. In the normal state of the body, the substance is practically not formed. Acids affect the functioning of the central nervous system. As a result, there is a disturbance in protein metabolism, as well as in the metabolism of lipoproteins and lipoproteins. At the same time, there are disorders in the transport of amino acids. The exchange of serotonin and catecholamines is disrupted. Perinatal factors become relevant.
Additionally, orthophenyl acetate and phenylethylamine begin to form. Normally they are also not produced. Possible in the presence of only minor amounts of precipitation. Excess leads to disruption of lipid metabolism. The process takes place in the brain. All this becomes the cause of a progressive pathology of the intellect. A person can even reach a state of idiocy. The final process of development of the pathology is not yet clear.
What does phenylketonuria look like with a photo?
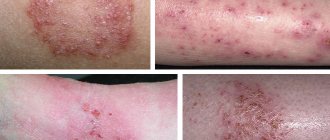
People suffering from pathology have a specific appearance. They always have very pale skin. She's practically white. This phenomenon occurs due to the fact that there is practically no pigment in it. The patient's hair is light-colored. Blue eyes. The skull may decrease slightly in size. Growth deviation from the norm is possible. Often it decreases a little. However, there are also examples of patients who had completely normal height. To better understand what a child with phenylketonuria looks like, it is recommended that you familiarize yourself with the photo.
The first signs of phenylketonuria
In newborn children who have the disease, no specific clinical manifestations are observed. The first signs of pathology can be noticed at the age of 2-6 months. During this period, the manifestation of the disease begins. As soon as milk protein enters the child’s body, the first signs begin to appear. They are non-specific. The child has:
- muscular dystonia;
- lethargy;
- hyperexcitability;
- constant regurgitation;
- convulsive syndrome;
- unmotivated anxiety.
One of the very first signs of the disease is persistent vomiting. However, it is often perceived as a clinical manifestation of pyloric stenosis. By 6 months of age, the child has a clear delay in psychomotor development. The patient becomes less active. He stops recognizing even the closest people. The child makes no attempt to sit down or stand on his feet. The composition of urine and sweat becomes abnormal. A characteristic mouse or moldy odor appears. Parents often notice the appearance of active peeling of the skin, dermatitis and eczema in their children.
If the patient does not undergo treatment, he will be diagnosed with microcephaly. Teeth will begin to emerge only after one and a half years, and developmental delays are clearly observed. By the age of three or four years, severe mental retardation and a complete lack of speech may occur. The only exceptions are some incomprehensible sounds.
Children with this disease have a dysplastic physique. They are often diagnosed with congenital heart defects. Autonomic dysfunction and chronic constipation may occur. Such a child is characterized by:
- mincing gait;
- tremor of the upper extremities;
- hyperkinesis;
- tailor's pose.
All of the above primary signs are characteristic of the classic form of the disease. With atypical types of pathology, increased excitability, tendon hyperreflexia and a high degree of mental retardation may be present. Gradually the disease progresses. By the age of two or three, the child may die.
Treatment with traditional methods
Due to the fact that phenylketonuria in children is considered a fairly serious disease, treatment with folk remedies is not carried out. It is possible to use medicinal plants to eliminate constipation, a characteristic sign of the disease, as sedatives and vitamin preparations.
You can take a medicinal decoction of mint, which has a calming effect and promotes better digestion of food.
You can take medicinal decoctions of mint, chamomile, calendula, lemon balm, hawthorn, motherwort, which have a calming effect on aggression and promote better digestion of food. Nuts, dried fruits with honey support the immune system. All prescriptions must be agreed with your doctor.
Treatment of the atypical form of the disease is not carried out due to its futility. Parents who are carriers of the mutated gene are monitored so that the newborn child is immediately tested for phenylketonuria and the necessary dietary therapy is prescribed in a timely manner, preventing the manifestation of a dangerous disease. Strict monitoring of families where children with pathology were born is also carried out.
Dietary treatment of phenylketonuria diagnosed at an early age is successfully carried out if the intake of animal protein into the patient’s body is strictly controlled. Over time, some patients recover completely and gradually switch to standard food. Some people use the diet for the rest of their lives. People who have had the disease are practically no different from their peers.
Symptoms of phenylketonuria
Symptoms of the disease in children with the classic form of pathology are not observed immediately after birth. However, such children have a number of specific external signs. They have:
- dryish white skin;
- pigmentation is almost completely absent;
- light-colored hair;
- Blue eyes.
At the age of 2 - 6 months, symptoms of the disease begin to appear. To the above symptoms are added lethargy, passive perception of the surrounding world, increased irritability and delayed psychomotor development. Sometimes frequent vomiting may occur. Unreasonable anxiety appears. Seizures may occur. If no pathologies are identified, protein foods are introduced into the child’s diet, and the symptoms begin to increase. The skull of such a child is somewhat reduced in size. Children suffering from pathology begin to walk later than their peers. In a year, such patients cannot express their emotions with their voice. They do not understand the speech of adults. Possible growth retardation.
Unconverted phenylalanine is excreted in sweat and urine, which leads to the appearance of a specific musty odor. The pathology also manifests itself in peculiar postures and gait. They arise due to the fact that the patient’s muscle tone is increased. In a standing position, the child spreads his legs wide and bends them at the knees and hip joints. At the same time, the head and shoulders are lowered. The gait is unsteady. The child takes small steps. Patients sit in the so-called tailor's position. They tuck and cross their legs. After 3 years, the child experiences increased excitability and fatigue. Behavioral and mental disorders are present. There is mental retardation. Very often the disease is accompanied by eczema, dermatitis and arrhythmia. If there is no treatment, the patient's condition will worsen. Timely diagnosis will reduce the impairments that the child may encounter.
Phenylketonuria (PKU) in children
New treatments
Today, researchers are developing several methods of alternative therapy for phenylketonuria (PKU):
- enzyme therapy with phenylalanine hydroxylase, phenylalanine ammonialyase
- "Large Neutral Amino Acids" Method
- treatment with tetrahydrobiopterin (Sapropterin)
There is information about cases where patients with moderate or mild forms of the disease were helped by tetrahydrobiopterin at a dose of 10 to 20 mg per 1 kg of body per day. In 2008, it was proven that for the normal physical development of children with phenylketonuria, dietary glycomacropeptides can be used, which also reduce the content of phenylalanine in the blood plasma and brain. An experimental method involves introducing the phenylalanine hydroxylase gene directly into the affected liver cells of a child. This method is not yet relevant in the CIS countries, including Ukraine and Russia.
Products for children with PKU (phenylketonuria)
Diet therapy helps prevent intellectual deficits in the classic form of the disease in question. The age of the baby is important when they start using the diet. Every month without diet, a child with PKU loses approximately 4 IQ points. The issue of diet for phenylketonuria in children is considered differently in different countries. But the principles are the same.
When the level of phenylalanine in the blood reaches 2–6 mg% (120–360 µmol/l) in infants, the diet is not used. The essence of nutrition for children with PKU is foods low in phenylalanine, mainly non-protein foods. Children of the first year of life need it. At a later age, such nutrition brings less results.
Therapeutic diet for PKU:
- natural food
- medicinal products
- low-protein starch-based products
Children with phenylketonuria should not:
- bird
- meat
- dairy
- fish
- breast milk (children up to 12 months)
Children with PKU only need formulas with minimal protein content. During the first year of life, the permissible amount of phenylalanine is from 90 to 35 mg/kg of the child. 50 mg phenylalanine = 1 g protein.
Medicinal products for phenylketonuria in children:
- XP Analogue LCP
- MD mil PKU-0
- Afenilac
The child must adhere to the diet if the phenylalanine level in the blood is at least 360–480 mmol/l.
Complementary feeding for phenylketonuria (PKU in children)
Once the child reaches 3 months of age, the diet should be expanded by introducing fruit and berry juices. At first it is 3-5 drops, then the volume is increased to 30–50 ml. For children 12 months old, the dose is already up to 100 ml.
Juices as complementary foods:
- pear
- apple
- plum
Fruit puree is also gradually introduced into the diet, gradually increasing the portion. Children from 4-4.5 months can already eat vegetable puree, which is prepared by their parents. You can also have canned fruits and vegetables for infants, but without milk. The second complementary food is porridge (10%) made from protein-free grains or sago. You can also give your child dairy-free, industrially produced cereals made from corn and/or rice flour. They should contain less than 1 gram of protein per 100 ml of finished product.
Children from 6 months of age can be introduced to the diet with jelly and/or mousses that do not contain protein. They are prepared with amylopectin swelling starch and fruit juice; These are the low-protein milk drink PKU “Loprofin” and the protein-free drink with milk flavor Nutrigen. Children with PKU from 7 months can be given low-protein products "Loprofin": rice, spaghetti, spirals. From 8 months, babies with phenylketonuria can be given special protein-free bread.
Diet for PKU in children over 1 year of age
To feed such patients, products based on mixtures of amino acids without phenylalanine and/or protein hydrolysates or with a negligible amount of it are used. They should contain complexes of macro-, microelements and vitamins. As the child gets older, the dose of protein can be increased, but not immediately. The amount of carbohydrates and fats should be gradually reduced, and then completely eliminated. The diet is gradually expanding to include natural products and dishes.
For children with PKU from 12 months, specialized medicinal products :
- Tetrafen 40
- Tetrafen 30
- MD mil PKU-1
- Tetrafen 70
- MD mil PKU-3
- Isifen
- P-AM 1, P-AM 2, P-AM 3
- XP Maxamum (neutral or orange flavor)
- XP Maxamade
Doctors advise gradually switching to products for older children over 1-2 weeks. The volume of the previous mixture should be reduced by 1/4–1/5 and an equivalent protein amount of the new product should be added. Some medicinal products contain polyunsaturated fatty acids. Among foreign-made low-protein products there are protein-free drinks, desserts, sauces and seasonings, cookies and special types of bread that children with phenylketonuria (PKU) can eat.
Some researchers are inclined to believe that children with PKU need to enrich their diet with tyrosine. Medicinal products have a specific taste, so flavoring additives without protein may be needed. You should not use the sweetener aspartame, because when broken down it also forms phenylalanine.
During treatment, regular monitoring of phenylalanine levels in the blood is necessary. Children under 3 months are tested once a week, and after stable results are obtained - at least once every 2 weeks. Children with PKU from 3 months. up to 1 year they check once a month, sometimes - 2 times. For children from 1 to 3 years of age, examinations are required at least once every two months, and after three years, monitoring is carried out once every 3 months.
It is necessary to monitor the following indicators for children with PKU:
- physical and intellectual development of the child
- nutritional status of the patient
- emotional development
- speech development
Once a month you need to conduct a general blood test. And according to indications - a biochemical blood test.
If a child is diagnosed with additional illnesses with dyspeptic symptoms, intoxication, hyperthermia, then the diet can be stopped for 2-3 days, replacing medicinal products with natural ones that do not contain too much protein. When the acute phase of the disease ends, medicinal products are reintroduced into the diet, but faster than at the beginning of the diet.
Termination of diet therapy
The age at which special diets should be stopped for PKU in children is still under debate. There is information that when diet therapy was discontinued at 5 years of age, one third of children with PKU experienced a decrease in IQ level by 10 points or more over the next 5 years. Discontinuation of the diet for children over 15 years of age in some cases affected progressive changes in the white matter of the brain, which was detected using MRI.
With classic phenylketonuria in children, the diet must be followed throughout life. The total amount of protein after adulthood should not be more than 0.8–1.0 g/kg per day.
Causes and prevention of phenylketonuria
The disease occurs due to a sharp decrease or absence of liver enzyme activity. It is with its help that phenylalanine is converted into tyrosine. Pathology occurs as a result of mutation of genes responsible for encoding enzymes. The damage affects the gene located on chromosome 12. Consanguineous marriages increase the occurrence of the anomaly. Diseases are inherited. Boys and girls encounter it equally often.
It is impossible to prevent the occurrence of the disease using preventive measures. Parents can only assess the risk of pathology in their child during pregnancy planning. To do this, you will need to consult with a geneticist, who will refer future parents to undergo specific studies. Particular attention must be paid to the process if there are consanguineous marriages, and if there were people in the family with this disease. If parents are carriers of the defective zvb gene, the risk of having a child with phenylketonuria is one in four.
Diagnostics
A blood test for phenylketonuria is carried out for each child in the maternity hospital.
The earlier a child is diagnosed with phenylketonuria, the better the prognosis and the greater the likelihood of normal intellectual development.
Due to the high prevalence of this pathology, the severity of the course and the real possibility of preventive treatment, WHO included it in the list of hereditary diseases for the detection of which mass screening of newborns is carried out. All children should be examined for phenylketonuria on the 4th-5th day of life. To do this, blood is drawn from the child, a special form is soaked with a drop of it, and it is sent for testing to the laboratory of the medical genetic center. Based on the results of this analysis (phenylalanine level), the child’s likelihood of developing the disease is assessed.
- When persistent hyperphenylalaninemia is detected, the classical form of the disease is differentiated from the atypical ones.
- With a slight increase in the concentration of this amino acid in the blood, repeated tests are performed.
At an older age, a doctor can assume the presence of a disease in a child based on clinical data. Children whose families have already reported cases of the disease have a particularly high risk of developing it. Laboratory tests can confirm the diagnosis:
- determination of the level of phenylalanine in the blood (more than 900 µmol/l);
- detection of its derivatives in urine (Felling test);
- detection of defective genes using molecular genetic methods (polymerase chain reaction).