Что такое «фенилкетонурия»?
Фенилкетонурия — врожденное генетическое заболевание, которое связано с нарушением обмена аминокислот. Вещество поступает в организм человека в составе белковой пищи. В результате происходит накапливание фенилаланина и его токсичных производных. В результате наблюдается поражение ЦНС. Патология характеризуется умственной отсталостью, когнитивными и поведенческими нарушениями. У детей наблюдается явное отсутствие желания и мысли сопоставлять всё вокруг и анализировать. Возможно появление пассивного интереса, но не более. Присутствует проблема с восприятием и адаптацией к социуму. Постепенно страдает и эмоциональная сфера. Наблюдается неустойчивость поведения.
В большинстве случаев развивается классическая форма заболевания. При ней наблюдается снижение активности печеночного фермента. Он носит название фениланин-4-гидроксилаз.
Существуют и другие разновидности патологии. В 1% случаев развивается атипичная разновидность заболевания. Она появляется в результате мутации другого типа генов. Он отвечает за процесс кодирования ферментов. Наследование заболевания осуществляется в соответствии с аутосомно-рецессивным схемой.
Из-за присутствия метаболического блока происходит активация побочных путей обмена фенилаланина. Это приводит к накоплению токсичных производных его действия. В частности, ими являются фениломолочная и фенилпировиноградная кислоты. При нормальном состоянии организма вещество практически не образуется. Кислоты оказывают влияние на работу ЦНС. В результате наблюдается нарушение белкового обмена, а также обмена липопротеидов и липопротеидов. Одновременно присутствуют расстройства в транспорте аминокислот. Нарушается обмен серотонина и катехоламинов. Актуальным становится перинатальные факторы.
Дополнительно начинают образовываться ортофенилацетат и фенилэтиламин. В норме они также не вырабатываются. Возможно в присутствии лишь незначительного количества осадков. Избыток приводит к нарушению метаболизма липидов. Процесс осуществляется в головном мозге. Всё это становится причиной прогрессирующей патологии интеллекта. Человек может достичь даже состояние идиотии. Окончательно процесс развития патологии пока не ясен.
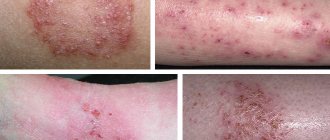
Как выглядит фенилкетонурия с фото
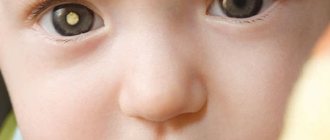
Люди, страдающие патологией, имеют специфическую внешность. У них всегда очень бледная кожа. Она практически белая. Подобное явление возникает из-за того, что какой-либо пигмент в ней практически отсутствует. Волосы пациента имеют светлый оттенок. Глаза голубые. Череп может незначительно уменьшаться в размерах. Возможно отклонение роста от нормы. Нередко он немного снижается. Однако имеются и примеры пациентов, которые обладали совершенно нормальным ростом. Чтобы лучше разобраться, как выглядит ребёнок с фенилкетонурией, рекомендуется ознакомиться с фото.
Первые признаки фенилкетонурии
У новорожденных детей, у которых присутствует заболевание, какие-либо специфические клинические проявления не наблюдаются. Первые признаки патологии можно заметить в возрасте 2-6 месяцев. В этот период начинается манифестация заболевания. Как только в организм ребёнка поступает белок молока, начинают возникать первые признаки. Они неспецифические. У ребенка наблюдаются:
- мышечная дистония;
- вялость;
- гипервозбудимость;
- постоянное срыгивание;
- судорожный синдром;
- немотивированное беспокойство.
Одним из самых первых признаков заболевания является упорная рвота. Однако его нередко воспринимают, как клиническое проявление пилоростеноза. К 6-месячному возрасту у ребенка наблюдается явное отставание в психомоторном развитии. Пациент становится менее активным. Он перестает узнавать даже самых близких людей. Ребёнок не предпринимает попыток сесть или встать на ножки. Состав мочи и пота становится аномальным. Появляется характерный мышиный запах или аромат плесени. Нередко родители замечают у детей появление активного шелушения кожи, дерматита и экземы.
Если больной не проходит лечение, у него будет выявлена микроцефалия. Зубы начнут прорезаться только после полутора лет, четко наблюдается задержка в развитии. К трем или четырем годам может возникнуть глубокая умственная отсталость и полное отсутствие речи. Исключение составляют лишь некоторые непонятные звуки.
Дети с таким заболеванием отличаются диспластическим телосложением. У них нередко выявляют пороки сердца врождённого характера. Возможно возникновение вегетативной дисфункции и хронических запоров. Для подобного ребенка характерно:
- семенящая походка;
- тремор верхних конечностей;
- гиперкинез;
- поза портного.
Все вышеуказанные первичные признаки характерны для классической формы заболевания. При атипичных разновидностях патологии может присутствовать повышенная возбудимость, сухожильная гиперрефлексия и высокая степень умственной отсталости. Постепенно заболевание прогрессирует. К двум-трем годам ребёнок может скончаться.
Лечение народными способами
В связи с тем, что фенилкетонурия у детей считается достаточно серьезным заболеванием, лечение народными средствами не проводится. Возможно употребление лекарственных растений для устранения запора, характерного признака болезни, в качестве успокоительных и витаминных препаратов.
Можно принимать лекарственный отвар мяты, который обладает успокаивающим действием, содействует лучшему перевариванию пищи
Можно принимать лекарственные отвары мяты, ромашки, календулы, мелиссы, боярышника, пустырника которые обладают успокаивающим агрессию действием, содействуют лучшему перевариванию пищи. Орехи, сухофрукты с медом поддерживают иммунную систему. Все рецепты требуется согласовывать с лечащим врачом.
Лечение атипичной формы заболевания не производится, в виду бесполезности. За родителями-носителями мутированного гена ведется наблюдение, чтобы новорожденного ребенка сразу проверить на фенилкетонурию, вовремя назначить необходимую диетотерапию, предупреждая проявление опасного заболевания. Также ведется строгий контроль семей, где родились дети с патологией.
Диетическое лечение диагностированной в раннем возрасте фенилкетонурии, успешно проводится, если строго контролировать поступление животного белка в организм пациента. Со временем часть пациентов полностью выздоравливают и постепенно переходят на общепринятую пищу. Некоторые используют диетическое питание до конца жизни. Люди, перенесшие заболевание, практически ничем не отличаются от своих сверстников.
Симптомы фенилкетонурии
Симптомы заболевания у детей с классической формой патологии сразу после рождения не наблюдается. Однако такие дети обладают рядом специфических внешних признаков. У них имеет место быть:
- суховатая кожа белого оттенка;
- пигментация отсутствует практически полностью;
- волосы светлого оттенка;
- голубые глаза.
В возрасте 2 — 6 месяцев начинают проявляться симптомы заболевания. К вышеуказанным признакам добавляется вялость, пассивное восприятие окружающего мира, повышенная раздражительность и задержка психомоторного развития. Иногда возможно возникновение частой рвоты. Появляется беспричинное беспокойство. Могут наблюдаться приступы судорог. Если патологий не выявлено, в рацион ребенка вводят белковую пищу, и симптоматика начинается нарастать. Череп такого ребенка несколько уменьшен в размерах. Дети, страдающие патологией, начинают ходить позже сверстников. В год такие пациенты не могут выразить голосом эмоции. Они не воспринимают речь взрослых. Возможна задержка роста.
Не преобразованный фенилаланин выходит с потом и мочой, что приводит к появлению специфического затхлого запаха. Патология проявляется также в своеобразных позах и походке. Они возникают из-за того, что мышечный тонус больного повышен. В положении стоя ребенок широко расставляет ноги и сгибает их в коленях и тазобедренных суставах. При этом голова и плечи опущены. Походка шатающаяся. Ребёнок делает мелкие шаги. Больные сидят в так называемой позе портного. Они поджимают и скрещивают ноги. После 3 лет наблюдается повышенная возбудимость ребенка и его быстрая утомляемость. Присутствует нарушение поведения и психические расстройства. Наблюдается умственная отсталость. Очень часто болезнь сопровождает экзема, дерматит и аритмия. Если лечение отсутствует, состояние больного ухудшится. Своевременная постановка диагноза позволит уменьшить нарушения, с которыми может столкнуться ребёнок.
Фенилкетонурия (ФКУ) у детей
Новые методы лечения
На сегодня исследователи разрабатывают несколько методов альтернативной терапии фенилкетонурии (ФКУ):
- энзимотерапия фенилаланингидроксилазой, фенилаланинаммониалиазой
- метод «больших нейтральных аминокислот»
- лечение тетрагидробиоптерином (Сапроптерин)
Существует информация о случаях, когда пациентам с умеренной или легкой формой заболевания помогал тетрагидробиоптерин в дозе от 10 до 20 мг на 1 кг тела в сутки. В 2008 году было доказано, что для нормального физического развития детей с фенилкетонурией можно применять пищевые гликомакропептиды, которые также снижают содержание в плазме крови и головном мозге фенилаланина. Экспериментальным методом считается введение непосредственно в пораженные клетки печени ребенка введение гена фенилаланингидроксилазы. Этот метод пока не актуален в странах СНГ, в том числе Украине и России.
Продукты для детей с ФКУ (фенилкетонурией)
Диетотерапия позволяет предотвратить интеллектуальный дефицит при классической форме рассматриваемой болезни. Важное значение имеет возраст малыша, когда начинают применять диету. Каждый месяц без применения диеты малыш с ФКУ теряет примерно 4 балла IQ. Вопрос о диете при фенилкетонурии у детей в различных странах рассматривают по-разному. Но принципы едины.
При уровне уровень фенилаланина в крови до 2–6 мг% (120–360 мкмоль/л) у младенцев диета не применяется. Суть питания детей с ФКУ – в продуктах с низким содержанием фенилаланина, в основном это небелковая пища. Она нужна детям первого года жизни. В более позднем возрасте такое питание приносит меньше результатов.
Лечебный рацион питания при ФКУ:
- натуральные продукты питания
- лечебные продукты
- малобелковые продукты на основе крахмала
Детям с фенилкетонурией нельзя:
- птицу
- мясо
- молочные продукты
- рыбу
- грудное молоко (детям до 12 месяцев)
Смеси детям с ФКУ нужны только с минимальным содержанием белка. В течение первого года жизни допустимое количество фенилаланина составляет от 90 до 35 мг/кг ребенка. 50 мг фенилаланина = 1 г белка.
Лечебные продукты при фенилкетонурии у детей:
- ХР Аналог LCP
- MD мил ФКУ-0
- Афенилак
Диеты ребенку нужно придерживаться, если показатель фенилаланина в крови составляет минимум 360–480 ммоль/л.
Прикорм при фенилкетонурии (ФКУ у детей)
По достижению ребенком 3-месячного возраста рацион нужно расширять, вводя фруктовые и ягодные соки. Сначала это 3-5 капель, потом объем увеличивают до 30–50 мл. Для детей 12 месяцев доза уже составляет до 100 мл.
Соки в качестве прикорма:
- грушевый
- яблочный
- сливовый
Также в рацион постепенно вводят фруктовое пюре, постепенно увеличивая порцию. Детям от 4-4,5 месяцев уже можно овощное пюре, которое готовится родителями. Также можно плодовоовощные консервы для грудничков, но без молока. Второй прикорм – каша (10%) из безбелковой крупки или саго. Также ребенку можно давать безмолочные каши промышленного производства из кукурузной и/или рисовой муки. В них должно быть меньше 1 грамма белка на 100 мл готового продукта.
Детям от 6 месяцев можно вводить в диету кисели и/или муссы, в которых нет белка. Их готовят на амилопектиновом набухающем крахмале и фруктовом соке; это низкобелковый молочный напиток PKU «Лопрофин» и безбелковый напиток с молочным вкусом Нутриген. Детям с ФКУ от 7 месяцев можно давать низкобелковые изделия «Лопрофин»: рис, спагетти, спиральки. С 8 месяцев при фенилкетонурии малышам можно давать специальный безбелковый хлеб.
Диета для ФКУ у детей от 1 года
Для питания таких пациентов применяют продукты на основе смесей аминокислот без содержания фенилаланина и/или гидролизатов белка или с мизерным его количеством. В их составле должны быть комплексы макро-, микроэлементов и витаминов. По мере взросления ребенка дозу белка можно увеличивать, но не сразу. Количество углеводов и жиров нужно постепенно снижать, а потом и вовсе исключить. Рацион постепенно расширяется за счет натуральных продуктов и блюда.
Для детей с ФКУ от 12 месяцев можно применять специализированные лечебные продукты:
- Тетрафен 40
- Тетрафен 30
- MD мил ФКУ-1
- Тетрафен 70
- MD мил ФКУ-3
- Изифен
- П-АМ 1, П-АМ 2, П-АМ 3
- ХР Максамум (вкус нейтральный или апельсиновый)
- ХР Максамейд
Врачи советуют постепенно переходить на продукты для детей более старшего возраста на протяжении 1-2 недель. Объем предыдущей смеси нужно уменьшить на 1/4–1/5 и добавить эквивалентное по белку количество нового продукта. В части лечебных продуктов содержатся полиненасыщенные жирные кислоты. Среди малобелковых продуктов зарубежного производства есть напитки без белка, десерты, соусы и приправы, печенье и специальные сорта хлеба, которые можно детям при фенилкетонурии (ФКУ).
Часть исследователей склоняется к мнению, что детям с ФКУ диету нужно обогащать тирозином. Лечебные продукты имеют специфический вкус, потому могут быть нужны вкусовые добавки без содержания белка. Нельзя применять подсластитель аспартам, потому что при расщеплении он образует в том числе фенилаланин.
При лечении нужен регулярный контроль содержания фенилаланина в крови. Детей до 3 месяцев проверяют 1 раз в неделю, а после получения стабильных результатов – минимум 1 раз в 2 недели. Детей с ФКУ от 3 мес. до 1 года проверяют 1 раз в месяц, иногда – 2 раза. Для детей от 1 до 3 лет осмотры нужны минимум 1 раз в два месяца, а после трех лет контроль проводят 1 раз в 3 месяца.
Необходим контроль таких показателей для детей с ФКУ:
- физическое и интеллектуальное развитие ребенка
- нутритивный статус больного
- эмоциональное развитие
- речевое развитие
Один раз в месяц нужно проводить общий анализ крови. А по показаниям – биохимический анализ крови.
Если у ребенка обнаруживают дополнительные болезни с диспепсическими явлениями, интоксикацией, гипертермией, то диету можно прекратить на 2-3 дня, заменив лечебные продукты на натуральные, в которых не слишком много белка. Когда острая фаза болезни заканчивается, в рацион снова вводят лечебные продукты, но быстрее, чем в начале ввода диеты.
Прекращение диетотерапии
Возраст прекращения специальной диеты при ФКУ у детей до сих пор в процессе дискуссии. Существует информация, что при отмене диетотерапии в 5-летнем возрасте у одной трети детей с ФКУ отмечалось снижение уровня IQ на 10 баллов и более на протяжении следующих 5 лет. Прекращения диеты для детей старше 15 лет в некоторых случаях сказывались на прогрессирующих изменениях белого вещества мозга, что было выявлено при помощи МРТ.
При классической фенилкетонурии у детей диеты нужно придерживаться всю жизнь. Общее количество белка после наступления совершеннолетия не должно быть больше, чем 0,8–1,0 г/кг в сутки.
Причины и профилактика фенилкетонурии
Заболевание возникает из-за резкого снижения или отсутствия активности фермента печени. Именно с его помощью происходит превращение фенилаланина в тирозин. Патология возникает в результате мутации генов, отвечающих за кодирование ферментов. Повреждения затрагивают ген, находящиеся на 12 хромосоме. Родственные браки повышают возникновение аномалии. Заболевания наследуются. С ним одинаково часто сталкиваются мальчики и девочки.
При помощи профилактических мер предотвратить появление заболевания нельзя. Родители могут лишь оценить риск появления патологии у ребёнка в процессе планирования беременности. Для этого потребуется пройти консультацию с генетиком, который направит будущих родителей на прохождение специфических исследований. Особое внимание необходимо уделить процессу, если имеет место быть кровнородственные браки, а также в роду присутствовали лица, болеющие этим заболеванием. Если родители являются носителzvb дефектного гена, риск появления ребенка с фенилкетонурией составляет один к четырём.
Диагностика
Анализ крови на фенилкетонурию проводят каждому ребенку еще в роддоме.
Чем раньше ребенку поставлен диагноз «фенилкетонурия», тем лучше прогноз и больше вероятность нормального развития интеллекта.
В связи с высокой распространенностью данной патологии, тяжестью течения и реальной возможностью профилактического лечения ВОЗ включила ее в список наследственных заболеваний, для выявления которых проводится массовый скрининг новорожденных. Все дети на 4-5-й день жизни должны обследоваться на фенилкетонурию. Для этого у ребенка производят забор крови, пропитывают ее каплей специальный бланк и отправляют на исследование в лабораторию медико-генетического центра. По результатам этого анализа (уровень фенилаланина) оценивают вероятность развития болезни у ребенка.
- При обнаружении стойкой гиперфенилаланинемии проводится дифференцирование классической формы болезни от атипичных.
- При незначительном увеличении концентрации этой аминокислоты в крови проводятся повторные анализы.
В более старшем возрасте предположить наличие заболевания у ребенка врач может на основании клинических данных. Особенно высокий риск его развития имеют дети, в семьях которых уже регистрировались случаи болезни. Подтвердить диагноз позволяют лабораторные исследования:
- определение уровня фенилаланина в крови (более 900 мкмоль/л);
- выявление его производных в моче (проба Феллинга);
- обнаружение дефектных генов с помощью молекулярно-генетических методов (полимеразно-цепная реакция).