Definition of disease. Causes of the disease
Fabry disease (Anderson-Fabry) is a rare genetically determined disease that is characterized by a decrease in the enzyme alpha-galactosidase A, which is responsible for the destruction of the sphingolipid globotriaosylceramide, the accumulation of which in the body leads to changes in the functioning of cells, progressive damage to the body, which is accompanied by a variety of clinical symptoms.
Included in the group of lysosomal storage diseases, sphingolipidoses.[3] The prevalence of Fabry disease ranges from 1 in 40,000 to 1 in 120,000 births.[4] The difference in data is explained by difficulties in making a diagnosis and different frequencies in populations.
In 1898, dermatovenerologist John Fabri (J. Fabri) first described a case of nodular purpura, subsequently complicated by macroalbuminuria in a thirteen-year-old boy.
The disease is caused by the presence of a mutation on the X chromosome. The mutation is in the GLA gene, located in Xq22.[3]
If you notice similar symptoms, consult your doctor. Do not self-medicate - it is dangerous for your health!
Fabry disease – enzyme deficiency
Fabry disease is caused by a decrease or absence of the enzyme alpha-galactosidase, caused by a mutation in the gene that controls its activity. With a lack of the enzyme, intermediate metabolic products accumulate in the lysosomes of the cell, and the metabolism of membrane glycosphingolipids is disrupted.
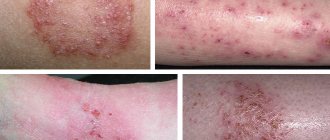
When metabolic products accumulate in small and large vessels, pathological processes gradually begin to develop in the body. The first signs are characteristic purple skin rashes, bouts of fever and severe pain in the arms and legs - the hands and feet are the first to be affected.
The only treatment available today is enzyme replacement therapy.
Treatment methods
The most effective and only method of treatment today is enzyme replacement therapy. This method allows the patient to replace an enzyme that is lacking in the patient’s body. The drugs began to be used successfully in the early 2000s. Taking them can significantly improve the patient’s well-being, but does not lead to complete healing.
Enzyme replacement therapy
For this purpose, recombinant alpha-galactose preparations are used. Today Replagal and Fabrazim are successfully used. Both are administered intravenously at regular intervals. The invasion process takes about half an hour. The drugs are very expensive, so treatment of patients with Fabry syndrome is paid for from the federal budget.
They help alleviate the patient’s condition and partially restore previously lost kidney function, reduce pain, stop the development of changes in the left ventricle of the heart, and prevent chronic renal and heart failure. Improvements in the patient's condition occur faster if treatment begins at the first symptoms of the disease.
Symptomatic treatment
To improve the patient’s well-being, drugs are used that can eliminate the pathological manifestations of the disease. To reduce pain and paresthesia, anticonvulsants, corticosteroids, analgesics and non-steroidal anti-inflammatory drugs are prescribed. To relieve pain, the use of patches and ointments containing lidocaine is recommended locally.
For kidney problems and arterial hypertension, ACE inhibitors and angiotensin II blockers are prescribed. In the thermal stage, hemodialysis or kidney transplantation is periodically performed.
In case of increased blood clotting, aspirin or cardiomagnyl are prescribed to prevent the formation of blood clots and thromboembolism, as well as to reduce the risk of stroke.
Angiokeratomas (spots on the skin) are removed using a laser.
Treatment
Various medications are used routinely to prevent cardioembolic events and ischemic strokes.
For this purpose, antiplatelet drugs are prescribed:
- Aspirin.
- Aspirin-dipyridamole.
- Clopedogrel.
- Ticlopidine.
- Warfarin.
Mental disorders can be corrected by prescribing:
- Carbamazepine.
- Haloperidol.
- Fentanyl.
- Phenytoin.
- Gabapentin.
Two enzyme preparations are used as pathogenetic therapy - Replagal and Fabrazim. They help normalize the function of the kidneys, heart and cerebrovascular tissues.
Surgery is indicated in late stages of the disease, when kidney or liver transplantation is necessary. However, it should be kept in mind that organ transplantation cannot slow down the development of FD in other systems.
Classification and stages of development of Fabry disease
There are two main forms of Fabry disease: [2]
- classical;
- atypical.
Classic is characterized by early manifestation of the disease (in the first decade of life), the presence of characteristic symptoms and complications.
In the atypical form, isolated damage to the brain (early strokes), heart and kidneys occurs; it manifests itself at a later age and poses great difficulties for diagnosis.
Some English-language authors also identify a female form of the disease. It has milder manifestations, beginning on average 5-10 years later than the classic “male” form.
Fabry disease: diagnosis of lesions in women
The following activities are implemented for females:
- Ormond's disease (retroperitoneal fibrosis): symptoms, diagnosis and treatment
- Pedigree analysis.
- DNA analysis.
- The level of the enzyme in the blood is not the main indicator of the disease, which is due to the asymmetric activation of the X chromosomes. It can be within normal limits, even if the woman has an illness.
Complications of Fabry disease
From the nervous system:
- ischemic strokes at a young age, often causing death.
From the cardiovascular system:
- ischemic heart attacks;
- cardiomyopathy;
- arrhythmias;
- heart failure leading to death.
From the kidneys:
- end-stage renal failure requiring kidney transplantation.
Other systems:
- visual impairment; hearing, up to deafness;
- bone fractures;
- overheating of the body.
Death most often occurs from uremia or ischemic damage to the brain and heart in the fourth decade of life.[8]
Symptoms
The disease, according to medical observations, first appears in adults and children during the pre- and puberty period.
The symptoms of the disease are as follows:
- Burning and soreness that occurs in the extremities, aggravated by physical activity and contact with heat. The pain is often accompanied by fever.
- Weakness. Very often, such manifestations are accompanied by a bad mood, anxiety, and a feeling of restlessness. Patients feel worsening mood and lack of self-confidence, which significantly reduces the quality of life.
- Decreased sweating.
- Fatigue of legs and arms. A person, as the disease develops, cannot perform even the most ordinary actions, as he experiences constant fatigue. Coupled with pain, such symptoms lead to complete helplessness.
- Proteinuria. The disease is accompanied by increased excretion of protein in the urine. Often this symptom causes an incorrect diagnosis.
- Formation of rashes in the buttocks, groin, lips, and fingers.
- Autonomic disorders.
Approximately a third of children develop articular syndrome simultaneously with the listed symptoms. The child experiences increasing muscle pain, decreased vision, and lack of cardiovascular activity. As enzyme deficiency progresses, kidney damage and increased blood pressure are observed. However, such a diagnosis is very rarely made to children, which is why it is so important to contact professional centers for blood testing at the genetic level in case of any manifestations.
Symptoms caused by the disease
Anderson's disease usually appears in childhood from 1 year of age. Adults get sick in rare cases. List of possible symptoms:
- irritation on the skin of the arms, legs, groin area and face, which appears in the form of ulcers; upon contact with hot objects, the affected area increases;
- cessation of sweat glands;
- severe weakness after light physical exertion;
- fever, increased temperature;
- the appearance of rheumatic joint syndrome;
- predominantly in children the appearance of vegetative disorders;
- muscle pain as the disease progresses;
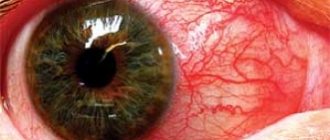
- loss of vision, cataracts may develop, the vessels of the retina are destroyed;
- heart pain;
- kidney problems;
- painful sensations when bending and straightening the limbs, which can last for several hours or weeks;
- after a heavy lunch and dinner, the stomach or intestines get sick;
- nausea;
- vomit;
- diarrhea.
Symptoms also include external manifestations of the disease. Usually external changes are noticeable at the age of 13 years. Typical characteristics in boys:
- retraction of the bridge of the nose;
- projection of eyebrows and forehead forward;
- 2 times enlarged lower jaw;
- the mouth doubles in size;
- lips widen.
Such changes are caused by somatotropic hormone.
The disease is accompanied by pain, which is divided into 2 types:
- Pain that occurs as a result of a physical stimulus is called neuropathic. Whenever you touch the feet and legs, the patient feels a burning and tingling sensation.
- Sudden pain. Appear suddenly in the form of attacks. Often the pain defect occurs on the feet and legs, sometimes in other parts of the body.
Diagnosis of Fabry disease
Early diagnosis is very important for correct and timely treatment of diseased organs and prevention of complications.[5]
An important method for diagnosing Fabry disease is to evaluate the patient's genealogical history, as this may reveal relatives with similar symptoms who are not aware of their disease. The gene responsible for Anderson-Fabry disease can be transmitted through a large number of generations, so many close and distant relatives are affected. To determine the risk of inheriting this disease, it is necessary to collect health information about all known family members.[3]
Fabry disease can be very difficult to distinguish from more common diseases, and patients may remain without a correct diagnosis for many years.
A preliminary diagnosis is made based on the results of a survey, patient complaints, and genealogical history.
To verify the diagnosis, methods for detecting substrates and enzymes and DNA diagnostics are used.
- Measuring the activity of alpha-galactosidase A. [2] In Anderson-Fabry disease, the activity of this enzyme in males is always below normal, and in females this indicator is within the normal range or slightly reduced. The materials for the study can be leukocytes, blood plasma, tear fluid, fibroblast culture.
- The most accurate diagnostic method is sequencing of the GLA gene .[2] To date, more than five hundred mutations of this gene leading to Fabry disease have been described. The use of this technique is limited by its cost. It is advisable to use DNA diagnostics for women, since the determination of alpha-galactosidase A activity in them does not always give a reliable result, and for relatives of the patient, since they may be carriers of a mutant gene or have an atypical form of the disease. This analysis can be carried out at the Center for Molecular Genetics in Moscow.
- Quantitative determination of globotriaosylceramide. This method has proven itself for monitoring the condition of patients and assessing the effectiveness of treatment, to determine the form of the disease (typical, atypical). The material for research can be blood plasma or dried blood spots.
- In some cases, a kidney biopsy to detect cells containing lysosomes with a characteristic substrate.
Differential diagnosis of Fabry disease is carried out with hereditary hemorrhagic telangiectasia, rheumatic fever, Fordyce disease, Schindler disease, and other hereditary storage diseases.[1]
Diagnostics
Diagnosis of Fabry disease is carried out on the basis of the clinical picture, as well as the results of laboratory and instrumental studies. When assessing the symptoms of pathology in men, a combination of signs such as:
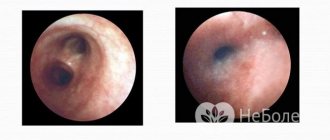
- angiokeratomas in the lower part of the body;
- chronic neuropathic pain in the extremities with periods of exacerbations (crises);
- corneal clouding;
- periodic increase in body temperature to febrile levels.
For laboratory tests, samples of blood (including dry spots), urine, tear fluid, and tissue can be used. Main research:
- Determination of alpha-galactosidase activity in leukocytes or skin fibroblasts using flowmetry. In men with Fabry disease, enzyme activity is reduced; in women it is close to the lower limit of normal.
- Kidney biopsy examination. Lysosomes with a characteristic substrate are detected in cells.
- Determination of the amount of sphingolipids in the blood. During illness, their number increases. The test is useful not only for making a diagnosis, but also for monitoring the patient's condition.
- DNA sequencing of the GLA gene is the most accurate method for detecting pathology. Its disadvantage is its high price.
If a genetic mutation is detected, it is advisable to carry out DNA diagnostics on all relatives of the patient who may have the same X chromosome.
Detection of Fabry disease in a child during embryonic development is possible by determining the activity of alpha-galactosidase in cells obtained from amniotic fluid or chorionic villi (fetal membrane). Such a diagnosis can only be carried out using invasive methods: amniocentesis, chorionic villus biopsy or cardiocentesis.
Instrumental methods used during examination for suspected Fabry disease:
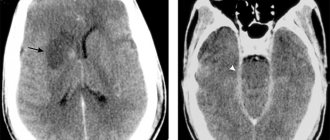
- MRI of the brain - demonstrates a hyperintense signal in the frontal and parietal lobes (in the white matter), damage to the posterior tubercle of the thalamus, vascular malformations;
- ECG - shows an increase (hypertrophy) of the left ventricle, a decrease in the P-R interval in the early stages and atrioventricular block in the terminal stage.
Fabry disease is differentiated from hereditary hemorrhagic telangiectasia (Randu-Osler-Weber syndrome) and rheumatism.
Features of the disease
What are the features of Fabry disease in men?
- Due to the fact that men have one X chromosome in their bodies, the likelihood of developing the disease is higher than in women.
- There is a pattern that a man whose body is affected by this disease cannot pass it on to his son. However, the incorrect gene is passed on to the daughters of the patient.
Features of Fabry disease in women:
- Women have two X chromosomes in their bodies. Therefore, the disease may occur in a milder form and its symptoms appear later.
- A woman can pass on the wrong gene to both her daughters and her sons. The probability that a child will inherit this disease is 50%.
Causes
The disease is based on a genetic defect in the sex X chromosome. In one of the sections of this chromosome, information about the enzyme α-galactosidase A is encoded. If a mutation occurs, this leads to a decrease in the amount of this enzyme or a decrease in its activity.
The defect has a recessive inheritance pattern. What does this mean? Since males have only one X chromosome (and the second is Y), boys with a pathological X chromosome always develop the classic picture of the disease. A sick man will definitely pass on the mutant X chromosome to all his daughters without exception (that is, in 100% of cases), but his sons will be healthy.
Women have two X chromosomes. If one of them is mutant, then such women experience clinical manifestations, but they are less pronounced, develop later and progress more slowly, almost always these are atypical forms of the disease. If a woman has both mutant X chromosomes, one from the father and the other from the mother (the probability of which is almost zero, given the prevalence of the disease), then the classic picture of Fabry disease also develops. A woman can pass on a mutant X chromosome to both sons and daughters (if there is one mutant X chromosome, the probability is 50%).
Treatment of Fabry disease
Treatment for Fabry disease involves replacing the deficient enzyme using enzyme replacement therapy. It is carried out using an intravenous infusion of the drug. Typically, enzyme replacement therapy is used in conjunction with various treatments for specific symptoms.[5]
In Russia today two drugs are used for enzyme replacement therapy: Fabrazyme, Genzyme; Replagal, Shayer. These drugs are very expensive; treatment of patients is paid for from the federal budget. Since the disease is classified as orphan (rare), there is a law on an accelerated procedure for researching drugs intended to treat such diseases.[3]
For men, initiation of enzyme replacement treatment is recommended as soon as possible after diagnosis.[5]
To relieve pain, drugs from the groups of anticonvulsants, antidepressants, analgesics, nonsteroidal anti-inflammatory drugs, and narcotic analgesics are used.[2]
Patients are advised to use antihypertensive drugs.
If end-stage renal failure develops, a kidney transplant is performed. Women who are relatives of the patient are not recommended to be donors.[2]
The use of hearing aids is indicated to correct hearing loss.
Physical activity in patients with Fabry disease should be limited due to the potential for increased symptoms and overheating.[3]
Situations in which it is inappropriate to prescribe enzyme replacement therapy:
- period of pregnancy and lactation;
- if there is another life-threatening disease in which the prognosis will not improve from the use of replacement therapy;
- there are serious complications (ischemic stroke, intensive care patients).
Forecast
Fabry disease has a relatively favorable prognosis if:
- timely administration of enzyme replacement therapy;
- regular monitoring of the patient’s condition (it is recommended to undergo a comprehensive medical examination at least once a year);
- following all doctor's recommendations.
Women with this disease are capable of childbearing. However, studies show that manifestations of pathology can provoke a number of complications both during the period of bearing a child and immediately after childbirth.
Without adequate treatment, Fabry disease negatively affects the quality of life of patients: they suffer from pain, heart and kidney problems, and decreased ability to work. Pathological symptoms negatively affect the psycho-emotional state of patients, and they develop depression.
The main causes of death in patients with Fabry disease are:
- renal failure;
- heart pathologies;
- stroke.
Average life expectancy: for men – 50 years, for women with a mild form of the disease – 50 years.
Fabry disease. Treatment and prevention
The patient is first prescribed painkillers. They alleviate the patient's condition. Another direction of treatment is to replenish the enzyme in the body that is not produced due to a damaged gene. It's called enzyme replacement therapy.
- Urolithiasis: causes, features of the course, diagnosis and methods of treating the disease
There is an opinion that if the disease is diagnosed at an early stage and this treatment is applied to the patient, Fabry disease can be overcome. There is no prevention for this disease, as it is inherited. The only way to avoid having a child with this disease is to test cultured amniotic cells for a-galactosidase A activity.
The best option for carriers of this disease is its early diagnosis. To do this, you do not need to let the child’s complaints about the presence of pain in the hands or feet take their course. Especially if there are known cases that one of the relatives in the family suffered from this disease. Modern medicine makes it possible to provide assistance to people who have Fabry disease in their bodies. Therefore, it is better to consult a specialist, do an examination of the body and begin timely treatment.
Fabry disease - what is it?
Lysosomal storage diseases are the general name for one large group of rare hereditary diseases that are caused by dysfunction of lysosomes. Fabry disease falls into this category. It is associated with a decrease in the activity of α-galactosidase, an enzyme in lysosomes that is responsible for the breakdown of glycosphingolipids. As a result, excess fats accumulate in cells and interfere with their normal functioning. As a rule, endothelial or smooth muscle cells of blood vessels, kidneys, heart, central nervous system, and cornea have to suffer.
Fabry disease - type of inheritance
This disease is considered genetically determined with an X-linked type of inheritance. That is, Fabry disease is transmitted only along the X chromosomes. Women have two of these, and therefore the anomaly can be inherited by both son and daughter. The probability of having a child with a genetic disorder in this case is 50%. Men have only one X chromosome, and if it is mutated, then Anderson Fabry disease will be diagnosed in their daughters with a 100% probability.
Fabry disease - causes
This is a genetic disease, and therefore the main reason for its appearance is mutational changes in the GLA genes, which are responsible for encoding the enzyme. According to statistics and the results of numerous medical studies, lysosomal Fabry storage disease is hereditary in 95% of cases, but there are exceptions. 5% of patients “earned” the diagnosis in the initial stages of embryo formation. This was due to random mutations.
Forecast. Prevention
With proper treatment and timely initiation of enzyme replacement therapy, the prognosis for patients is favorable. If untreated, death from cardiovascular, neurological and renal complications occurs in the fourth decade of life.[4]
Prevention of the disease consists of prenatal or preimplantation (with IVF) diagnosis of the presence of a mutant gene using DNA diagnostics and termination of pregnancy for medical reasons.[4] It is necessary to examine all potential carriers of the gene in order to promptly prescribe enzyme replacement therapy and prevent clinical manifestations of the disease and its complications. A screening study of dried blood spots with determination of alpha-galactosidase activity is possible, but it is limited by the financial capabilities of the region. It is essential that all patients with signs of Fabry disease be tested for this disease. For this purpose, there is a program regulating the collection and sending of material to the Center for Molecular Genetics, and the provision of patients with replacement therapy drugs.
Diagnosis of the disease in men
The following events are held for males:
- Analysis of pedigree, family tree. Given the hereditary nature of the lesion, the collection of a family history is of primary importance in diagnosis. But in members of the same family, the disease may remain unrecognized.
- Test for the level of galactosidase enzyme in the blood.
- DNA analysis is carried out in case of ambiguous results of biochemical studies. DNA can be taken from any biological material.
Bibliography
- Kozlova S.I., Demikova N.S. Hereditary syndromes and medical genetic counseling. M: T-vo scientific publications KMK; Author's Academy, 2007. 448 p.
- Federal clinical guidelines for the diagnosis and treatment of Fabry disease. Moscow 2013.
- Information about Fabry disease (date of access: 01/25/18)
- Mehta A, Ricci R, Widmer U, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. European Journal of Clinical Investigation 34(3):236–42
- Union of Patients Suffering from Fabry Disease ((access date 01/25/18)
- Omid Motabar, Ellen Sidransky, Ehud Goldin, and Wei Zheng. Fabry Disease – Current Treatment and New Drug Development. Curr Chem Genomics. 2010; 4:50–56
- Stephen Waldek, Sandro Feriozzi. Fabry nephropathy: a review – how can we optimize the management of Fabry nephropathy? BMC Nephrol. 2014; 15:72
- Frank Weidemann, Maria D Sanchez-Niño et al. Fibrosis: a key feature of Fabry disease with potential therapeutic implications. Orphanet J Rare Dis. 2013; 8:116
- Juan M. Politei, Didier Bouhassira et al. Pain in Fabry Disease: Practical Recommendations for Diagnosis and Treatment. CNS Neurosci Ther. 2020 Jul; 22(7): 568–576
Diagnosis of the disease
Correct diagnosis often causes difficulties, since the disease is quite rare and manifests itself in a whole range of clinical symptoms. Patients suspected of having Fabry disease are examined by specialists in various clinical areas.
Practical experience shows that on average, about 12 years pass between the initial manifestations of symptoms and the final diagnosis. It is very important to identify the disease as early as possible in order to begin full treatment.
Health care providers should suspect the presence of this genetic disorder if a person has a combination of more than two symptoms of the disease. In early childhood, Fabry disease (a photo of the affected chromosome is presented below) practically does not manifest itself, so diagnosis can be complicated.
Complications
With further progression of Fabry disease, the following complications may occur:
- Urolithiasis symptoms and treatment in men
- Kidney function is destabilized. Protein begins to be excreted from the body. Kidney failure develops.
- Damage to the cardiac system. Its shape may change and arrhythmia may occur.
- Inadequate nutrition to the brain, which increases the likelihood of a stroke.
How does it manifest?
The peculiarity of this disease is that it has different symptoms in different people. Some people have mild symptoms throughout their lives. And for some they manifest themselves in severe forms. As mentioned above, in representatives of the stronger sex, signs of the disease can be noticed earlier than in girls.
Statistics show that the diagnosis of Fabry disease is made to people between the ages of 30 and 45 years. Although the person had signs of the disease before.
This should be paid attention to if there is a possibility of inheritance of this disease.
Sources used:
- https://probolezny.ru/bolezn-fabri/
- https://onevrologii.ru/nasledstvennye-zabolevaniya/bolezn-fabri
- https://fb.ru/article/177927/bolezn-fabri-simptomyi-lechenie-foto
- https://www.syl.ru/article/292490/bolezn-fabri-prichinyi-simptomyi-lechenie
Fabry disease - symptoms
Signs of the disease manifest themselves differently in different organisms:
- Men.
In representatives of the stronger sex, Anderson-Fabry disease, as a rule, begins to manifest itself in childhood. The first signs: pain and burning in the limbs. Some patients complain of a purple rash, which in most cases covers the area from the navel to the knees. Since the disease progresses slowly, serious symptoms - discomfort in the abdomen, ringing in the ears, frequent urge to defecate, pain in the back and joints - become noticeable only by the age of 35 - 40. - Women.
In the female body, the disease shows a wide range of clinical manifestations. While some patients are unaware of their problem, others suffer from corneal dystrophy, fatigue, cardiovascular disorders, anhidrosis, gastrointestinal disorders, kidney disease, eye damage, and neurological disorders. - Children.
Although in most cases the first symptoms of the disease appear early, Fabry disease in children often goes unnoticed and develops until adulthood. The earliest signs are pain and angiokeratomas, which are often located behind the ears and are overlooked by specialists. Other manifestations of the disease in young patients: nausea with vomiting, dizziness, headaches, fever.