Causes
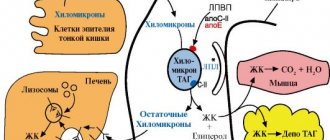
Pompe syndrome is caused by a mutation in the GAA gene, which is located on chromosome 17. This gene is responsible for encoding the lysosomal enzyme acid alpha-glucosidase. It is an important catalyst for the breakdown of the glycogen molecule into glucose, which is involved in the energy metabolism of cells. In other words, there is a disruption of glycogen metabolism, which causes a lack of energy for muscle and nerve fibers, liver, and myocardium.
Dystrophy and cell damage also occur due to the accumulation of glycoside in lysosomes. Inheritance of the GAA gene occurs in an autosomal recessive manner, i.e. the patient receives the mutant allele from both parents. The form of the disease affects the choice of treatment and further prognosis.
Classification
Pompe disease is rare in adults. Children are mainly susceptible to glycogenosis. The timing of the development of the disease and its symptoms determine the forms of the disease:
- Early – manifests itself at birth or in the first months of life, and is considered the most dangerous form of the disease (mortality rate is 100% before the age of 1 year).
- Late – symptoms are observed at 1-3 years. Partial disappearance of alpha-glucosidase or a decrease in its activity determines the late form of Pompe syndrome.
- Juvenile – development of characteristic features at 6-10 years of age.
- Adult – the development of symptoms occurs in the range of 20-40 years. Patients experience slowly progressive myocardial disorders.
Pompe disease: together we are strong
On April 15, patient organizations and patients with Pompe disease around the world are conducting education and awareness campaigns to raise awareness among the general public about this rare inherited disease with an autosomal recessive mechanism of inheritance.
Child with Pompe disease
The exact prevalence of Pompe disease is unknown. According to various authors, the frequency of the disease, depending on the country and ethnicity, varies from 1:40,000 to 1:300,000.[1]With this diagnosis, as a result of a lack of a certain enzyme, a special substance glycogen accumulates in the cells of the patient’s muscles and heart[ 2].
In families where parents are carriers of Pompe disease, the risk of having an affected child is 25% for each pregnancy.[3] If one child is diagnosed with this, this does not mean that all children will be sick, but it is necessary to examine all brothers and sisters.
A characteristic symptom of the disease is progressive muscle weakness, including skeletal and respiratory muscles[4]. Damage to the heart muscle manifests itself mainly at the onset of the disease at a very early age. This form of the disease is called infantile. It progresses rapidly and leads to death in the first year of life.5 In cases of juvenile or adult forms, progression of the disease includes increasing weakness of the muscles of the trunk and legs, gait disturbance, shortness of breath, respiratory failure, difficulty chewing and swallowing, due to rapid fatigue of the masticatory muscles . Without therapy, over time, the symptoms of the disease increase. Many patients stop walking independently and gradually become dependent on a ventilator as the muscles responsible for breathing become damaged. [5], [6]
Unlike many other rare diseases, enzyme replacement therapy is available for patients with Pompe disease, the effect of which largely depends on when the patient began receiving treatment. With timely and regular supply of the missing enzyme, the progression of the disease can be stopped or slowed, allowing people with this disease to lead an active social life.
Early diagnosis plays a key role in the treatment of Pompe disease.
“When diagnosing rare diseases in general and Pompe disease in particular, primary care physicians have a great responsibility. It is very important that the doctor suspects a “rare diagnosis” in time and refers the patient for examination if necessary. But the patient himself must also be quite persistent and at the same time have patience in order to find the cause of the disease and possibly conduct an examination of the whole family. You need to understand that there is no “orphan” doctor, there is a doctor who wants to figure it out. On average, a patient with a rare disease must see about 7 doctors before being diagnosed. And this is not only in Russia, it is all over the world!” emphasized E. Yu. Zakharova, head of the laboratory of hereditary metabolic diseases of the Medical Genetic Research Center of the Russian Academy of Sciences, Doctor of Medical Sciences.
“We noticed the first changes in our daughter’s body during a medical examination for VHI. The process of making a diagnosis was long, notes E., the mother of a patient with Pompe disease. “At first they diagnosed undifferentiated hepatitis, but the treatment did not help. Then they found a doctor who helped make the diagnosis - Pompe disease. In total, we tried to establish a diagnosis for a year and a half. Afterwards, a long period of accepting the diagnosis began, studying information and adapting to life with the disease.”
In Russia, a disease is considered rare (orphan) if its prevalence is no more than 10 cases per 100,000 population.[7] Most often they are genetic and appear in childhood7, but the onset of the disease can also occur in adulthood. With adequate treatment and systematic use of medications, people with orphan diseases can lead active lives and be full-fledged members of society.[8]
[1] https://www.pediatr-russia.ru/sites/default/files/file/kr_pompe.pdf
[2] Hirschhorn, Rochelle and Arnold JJ Reuser. Glycogen Storage Disease Type II: Acid Alphaglucosidase (Acid Maltase) Deficiency. In: Scriver C, Beaudet A, Sly W, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th Edition. New York: McGraw-Hill, 2001. 3389-3420.
[3] https://pompe.rare-diseases.ru/
[4] Kishnani PS, Steiner RD, Bali D, et al. Pompe disease diagnosis and management guideline. Genet Med 2006; 8:267-88
[5] Van den Hout HM, Hop W, van Diggelen OP, et al. The natural course of infantile Pompe's disease: 20 original cases compared with 133 cases from the literature. Pediatrics 2003; 112:332-40.
[6] Van der Beek Naet al. Clinical features and predictors for disease natural progression in adults with Pompe disease: a nationwide prospective observational study. Orphanet J Rare Dis. 2012 Nov 12;7:88
[7] https://spiporz.ru/redkie-zabolevaniya/
[8] https://philanthropy.ru/intervyu/2016/03/11/35529/
Symptoms
The characteristic signs of a genetic disease depend on the form of glycogenosis. The symptoms are:
| Early | Late and Juvenile | Adult |
| Breathing disorder | Muscle atrophy (up to the diaphragm) | Increasing muscle weakness |
| Bloating | Problems with motor coordination | Morning headache |
| Muscle hypertrophy upon palpation | Late start of walking | Drowsiness or insomnia |
| Cardiomyopathy | Dysphagia (swallowing problems) | Scoliosis of the thoracic region |
| Changing the size of the tongue (increase) | Respiratory dysfunction | Shortness of breath on exertion |
| Swallowing disorder, sluggish sucking | Frequent colds, ARVI | Orthopnea (impaired breathing in a horizontal position) |
| Enlarged liver, heart | Hepatosplenomegaly (enlarged spleen, liver) | Signs of heart failure |
| Arrhythmia (irregular heart rhythm) | Stunting |
Articles on the topic
- Solutions for inhalation with a nebulizer for bronchitis for children and adults with a description
- Diseases of the lower extremities - signs, classification, diagnosis, therapy and prevention
- Myopathy - what is it in children and adults
Manifestations of late infantile and juvenile forms
In late and juvenile forms of the pathology, the clinical picture is very similar. The difference lies only in the timing of the appearance of the first signs, namely the identification of muscle weakness. Later, the symptoms are supplemented by disturbances in the functioning of the heart muscle. A few months after the first signs appear, doctors diagnose the formation of muscular dystrophy of skeletal muscles and cardiomegaly.
The duration of the disease in each specific case may vary, but the average period is 9-12 years. In this case, indirect symptoms of the pathology may appear in the form of headaches, sleep apnea, and frequent respiratory diseases of varying complexity. Lack of therapy leads to death due to heart failure.
Diagnostics
Several diagnostic methods are used to identify Pompe syndrome. The initial stage of diagnosis requires the implementation of general procedures:
- family history analysis;
- determination of the main symptoms through examination, palpation, data collection;
- laboratory tests (general and biochemical blood tests, genetic tests, PCR tests);
- MRI, CT, ECG, EchoCG;
- chest x-ray;
- Ultrasonography of the abdominal cavity – obtaining images of organs and tissues.
Clarification of the diagnosis requires the use of specific diagnostics. Her methods include:
- Amniocentesis is a study of amniotic fluid for genetic abnormalities, carried out during the perinatal period.
- Establishing the degree of activity of acid alpha-glucosidase is carried out by checking leukocytes, as well as dried blood spots.
- DNA research is the study of the GAA gene for mutations.
- A biopsy of the affected muscle tissue reveals the presence of glycogen accumulation and cavities in the cytoplasm.
The late form of Pompe syndrome is diagnosed using certain methods. These include:
- Polysomnography is a study of blood counts during sleep to determine apnea.
- Electromyography of skeletal muscles is an assessment of the electrical activity of muscle mass.
- Establishing vital capacity of the lungs - determining damage to the diaphragm using the difference in indicators while standing and lying down.
Diagnostics[edit | edit code]
The diagnosis of this rare hereditary pathology is verified on the basis of a laboratory study of the degree of activity of the enzyme - acid α-1,4-glucosidase (maltase) in the blood (dry spot method) [7] or body tissues (skin fibroblasts, muscle tissue, leukocytes).
The disease occurs in various clinical forms:
- classic (infantile) form of Pompe disease and
- Late-onset Pompe disease (in childhood, adolescence and adults).
Stages of diagnostic search[edit | edit code]
Classic (infantile) form:
- stage: identifying the risk group.
- main clinical manifestations: severe muscle hypotonia (“floppy child” syndrome);
- delayed motor development;
- frequent infectious diseases of the upper respiratory tract (ARD);
- presence of signs of respiratory failure;
- breathing disorders during sleep;
- cardiomegaly in combination with cardiomyopathy;
- heart failure;
- macroglossia;
- hepatomegaly;
- splenomegaly;
- malnutrition
- laboratory diagnostics: increased levels of creatine phosphokinase, lactate dehydrogenase, ALT, AST in the blood serum.
- instrumental diagnostic methods:
- searching for signs of myocardial damage and/or heart rhythm;
- ECG;
- EchoCG;
- MRI;
- X-ray of the chest organs
- stage: specific diagnosis.
- diagnostics: for all patients at risk of Pompe disease - biochemical analysis of the activity of acid maltase in the blood using the dry spot
- verification: repeated measurement of the level of acid maltase activity in the blood using an alternative method (for example, determination of the activity of this enzyme in peripheral blood leukocytes)
- stage: additional diagnostic methods.
- in the presence of a typical clinical picture against the background of “normal” activity of acid maltase in the blood, DNA diagnostics of the defective gene (GAA) is indicated.
- morphological analysis of muscle tissue (biopsy) reveals specific changes, but their absence does not exclude the presence of Pompe disease.
Diagnostic algorithm for late-onset Pompe disease:
- stage: identifying the risk group.
- main clinical manifestations: progressive muscle weakness of the pelvic and shoulder girdle (in the initial stages of the disease, weakness in the legs predominates);
- difficulty walking, climbing stairs, getting up from a sitting position;
- muscle pain;
- respiratory failure;
- orthopnea;
- frequent infectious diseases of the respiratory tract;
- daytime sleepiness;
- headache in the morning;
- nocturnal hypoventilation of the lungs;
- weight loss in children and adolescents;
- malnutrition
- laboratory diagnostics: increased levels of creatine phosphokinase, lactate dehydrogenase, ALT, AST in the blood serum.
- instrumental diagnostic methods:
- identification of a myopathic pattern during myography of skeletal muscles of the limbs;
- detection of breathing disorders during sleep (episodes of apnea and decreased blood oxygenation) during polysomnography;
- identification of a significant difference in the results of measuring forced vital capacity (FVC) in the supine and sitting position by more than 10% from the initial FVC value - an early sign of specific damage to the diaphragm in Pompe disease.
- stage: specific diagnosis.
- diagnostics: for all patients at risk of Pompe disease - biochemical analysis of the activity of acid maltase in the blood using the dry spot
- verification: repeated measurement of the level of acid maltase activity in the blood using an alternative method (for example, determination of the activity of this enzyme in peripheral blood leukocytes)
- stage: additional diagnostic methods.
- in the presence of a typical clinical picture against the background of “normal” activity of acid maltase in the blood, DNA diagnostics of the defective gene (GAA) is indicated.
- morphological analysis of muscle tissue (biopsy) reveals specific changes, but their absence does not exclude the presence of Pompe disease.
Treatment
Pompe syndrome cannot be completely cured. Specific therapy is considered to be lifelong use of enzyme replacement treatment. It is carried out using recombinant acidic alpha-glucosidase. The substance helps improve the patient’s quality of life, slow the progression of symptoms, and reduce the risk of complications.
In Russia, the only available drug of this group is Myozyme. It is used intravenously to restore affected organs and systems. The use of a recombinant enzyme reduces mortality by 99% and the risk of respiratory complications by 92%. The drug is prescribed for continuous use.