Craniostenosis is a brain disease that develops as a result of craniosynostosis, that is, premature fusion of the sutures between the bones of the skull or due to their congenital absence. This disrupts the healthy correspondence between the growth of the brain and the bones of the skull. The result of such a violation is deformation of the skull and a serious increase in intracranial pressure. The incidence of the disease in children is 1:1000 – 1:1900. Treatment of craniostenosis requires surgical intervention.
Diagnostics
What parents should pay attention to
- Unusual shape of a child's head
- Early closure of the large fontanel (up to a year), discrepancy between the growth rate of the child’s head circumference and the age norm (see head circumference of boys and girls)
- Poor sleep, restlessness of the child, deterioration of the child when the weather changes, regurgitation, lag in psycho-motor development (see Assessing the level of psychomotor development of the child)
If the above symptoms are detected in a child, you should contact a specialist:
- Neuropathologist – assesses the presence of neurological symptoms and delayed development of the child
- Ophthalmologist – assesses signs of intracranial hypertension based on the results of fundus examination (in advanced cases – decreased visual acuity)
- Pediatrician – assesses the presence of other anomalies in the development of organs and systems, somatic pathology
- Genetics – reveals the presence of the genetic nature of the disease, the likelihood of anomalies in other organs and systems and the prognosis of the recurrence of a similar pathology in the next child
It is necessary to carry out instrumental examination methods:
- Craniometry (cephalometry) – measurement of standard and special anthropometric indicators of the skull, indices. It is carried out using measuring instruments (centimeter tape, cephalometer) and using special computer programs based on Sp-CT studies.
- Ultrasound of internal organs for developmental anomalies.
- ECHO - cardiography - reveals abnormalities in the development of the cardiovascular system (open oval window, additional chords in the cavities of the heart)
- ECG – detects heart rhythm disturbances
- Ultrasound functional venography of the main vessels of the head and neck - reveals a violation of the venous outflow of blood from the cranial cavity and allows you to indirectly assess and identify signs of intracranial hypertension.
To confirm the diagnosis, Spiral Computed Tomography (Sp-CT) is performed - it reveals premature closure of the skull suture, the degree of intracranial hypertension. Allows you to determine the indication for surgery and its planning.
Important! In most cases, the examination must be performed under sedation so that the child can tolerate the examination calmly. It is necessary to take into account that sedation is carried out on an empty stomach and the last feeding before the introduction of an anesthetic should be at least 3 hours before.
With the results of examinations (conclusions), Sp-CT images, you must contact a Neurosurgeon
Based on the results of all the above studies, a decision is made on an operation, the purpose of which is 1. a vital increase in the volume of the skull for the full development of the child’s brain and 2. cosmetic correction of the abnormal shape of the skull.
In the case of complex and syndromic forms of craniostenosis, it is necessary to conduct additional examination methods, such as – Angiography, which allows identifying abnormal blood supply pathways to the brain and skull and blood outflow from the cranial cavity in order to plan optimal surgical tactics and reduce operational risk.
Joint discussion and planning of surgical intervention with the participation of maxillofacial surgeons is mandatory,
Making a three-dimensional model of the skull using stereolithography
When the diagnosis of “Craniostenosis” is confirmed and the decision to undergo surgery is made, the parents’ next step is: Hospitalization
There are many options for surgical intervention for this pathology, each of which is used depending on the nature of craniostenosis. At the same time, surgical technology does not stand still; recently, resorbable materials have become widely used, which make it possible to create a strong frame that holds the volume and shape of the skull newly created surgically for 6 months - 1 year, the time required for the formation of one’s own bone, followed by resorption of these materials.
The duration of the operation depends on the type of craniostenosis, the age of the child, the presence of concomitant pathology (which does not make surgical intervention contraindicated) and developmental anomalies of other organs and systems and usually ranges from 3 hours.
After the operation, the child is transferred to the general department within 24 hours, where he stays with his mother or father. As a rule, the patient is observed in the department for 8 days, after which the stitches are removed and the baby is discharged home.
After discharge home
The child must be observed by the operating surgeon for several years after the operation. The observation period depends both on the form of craniostenosis and on the age at which it was operated on.
The first examination after surgery is usually carried out after 3-6 months. Sp-CT must be performed before consultation
It is quite difficult to detect craniostenosis in utero. But there is a chance that a standard ultrasound examination of the fetus (in the third trimester) of pregnancy may reveal deformation of its head, especially if a 3D ultrasound scan is performed. Deformation of the skull in the fetus and suspicion of craniostenosis may be an indication for cesarean section to avoid injury to the birth canal of mother and child. After all, with craniostenosis, the bones will not be able to form correctly during childbirth.
— First of all, you need to pay attention to any changes in the shape of the skull. An elongated skull, a sloping forehead or back of the head, a ridge in the forehead, flattening of the child’s head in the anteroposterior direction, the presence of a bone ridge behind the large fontanel above the fused sagittal suture, close or vice versa, wide-set or asymmetrically located ocelli, protrusion of the ocelli from the eye sockets, symmetry of the ears.
A large fontanel may also close early, or vice versa, remain open at an older age (with syndromic forms). Normally, a large fontanel in a baby closes before one year, even up to one and a half years. If the fontanel closed much earlier or, moreover, the baby was born with a closed fontanel, contact a neurosurgeon for advice.
— Is craniostenosis always associated with an abnormal development of the brain itself?
- Not at all necessary. Yes, it happens that we encounter microcephaly - a primary brain abnormality in which the skull does not grow because the brain does not develop. The brain does not grow - there is no impetus for the development of the skull bones, the sutures and fontanel close prematurely. In such a situation, unfortunately, surgical intervention cannot help.
— At what age are such operations performed?
- It depends on the type of craniostenosis. For scaphocephaly, the optimal age for surgery is up to 6 months. In this case, endoscopic surgery can be performed. If parents contact us later than this period, then it is no longer endoscopic, but ordinary reconstructive surgery. It is usually carried out between the ages of 1 year 2 months and 1 year 5 months.
If a child is not diagnosed with craniostenosis on time, but the deformity is discovered, for example, at the age of 3 years, operations, as a rule, are no longer performed. The brain has already formed under unfavorable conditions, and it is more difficult to obtain a good cosmetic result from changing the shape of the skull.
— What does a neurosurgeon do during an operation?
— With scaphocephaly, for example, the surgeon endoscopically excises the bone in the area of the synostotic sagittal suture and makes “notches” on the sides, on the parietal bones, so that the parietal bones can diverge. If the child wears a special helmet after surgery, a normal skull will form earlier.
The anteroposterior size of the skull will be limited in height by the helmet, and the parietal bones will be able to diverge to the sides. For other forms of craniostenosis, different tactics are used. For example, with trigonocephaly (a ridge in the forehead, a triangular shape of the skull), the frontal bone is reconstructed, the distance between the eyes increases (with trigonocephaly it is narrowed), and the frontal region is advanced. Due to this, the volume of the skull increases, the shape and proportions approach the norm.
— Is surgical intervention indicated in all cases of early diagnosis?
— Craniostenosis is a fairly rare pathology.
Much more often we encounter positional deformation of the skull. This is the scourge of our time.
Parents now have many restraint devices such as car seats at their disposal. And they often leave the child in them lying in one position for a long time. Or, say, a child lies in a crib and looks all the time towards the working TV. This is convenient for mom, he requires less of her attention. But the longer he lies, the more the back of his head flattens on one side.
— If we rule out a brain abnormality and perform surgery for craniostenosis in time, can we hope that the child will develop fully?
- Yes. Certainly. This is why the operation is performed. For craniostenoses, with the exception of syndromic ones, preventive surgery is performed to avoid problems that may arise in the future. We subsequently monitor such patients for 2-3 years. And then the need simply disappears. They don't contact us anymore.
Diagnosis of craniosynostosis is based on physical examination and instrumental research methods. The anamnesis is often uninformative, but its data allows the pediatrician to trace the dynamics of clinical symptoms, if any. An important point is a visual examination of the child, which makes it possible to detect characteristic deformations of the skull, bone anomalies, etc. Laboratory tests do not reveal specific changes and can be used to determine genetic pathology or diagnose complications.
Instrumental methods are required to visualize bone deformations and assess the degree of damage to brain tissue. This includes neurosonography, radiography, computed tomography and magnetic resonance imaging. Neurosonography is used to assess the condition of brain tissue and the size of the ventricles, and to identify intracranial hypertension. An x-ray can detect abnormalities in bone structure, ossification of cranial sutures, and, with increased intracranial pressure, increased digital impressions. CT and MRI are used to obtain more informative results. If damage to the visual system is suspected, ophthalmoscopy is performed to detect damage to the optic nerve head. Consultations with a neurosurgeon and ophthalmologist are recommended.
Differential diagnosis of craniosynostosis is carried out with positional plagiocephaly, birth trauma of newborns (cephalohematoma, subgaleal hemorrhage, skull fracture), brain cysts, rickets and microcephaly.
If the baby's head is too small
If there are disturbances in the structure of the bony skeleton of the skull, the baby's head may be too small in size and a serious disease is detected - craniostenosis. This is an abnormally early fusion of cranial sutures, which affects approximately 1 in 2000 newborns. As a result of this abnormal ossification, the baby's head takes on an abnormal shape, which limits brain growth and normal circulation of cerebrospinal fluid. Various symptoms may then occur - unexplained bouts of vomiting, visual disturbances, hearing loss and mood disturbances, changes in mood and frequent crying.
If the baby's head does not develop properly, the most important brain structures suffer, which leads to disruption of neuropsychic development, leading to mental retardation and disability, and sometimes to the death of children. There are many variants of this pathology, it all depends on which of the cranial sutures fuse ahead of time. Total fusion of all sutures on the skull is very rare; usually several of them are affected, while others grow quite normally and close on time. Then the baby’s head takes on a special shape; the pathology may be accompanied by fusion of the vertebrae, respiratory dysfunction, strabismus, or even complete blindness.
Causes of craniosynostosis
The exact etiology of craniosynostosis has not been established. According to the theories put forward, this disease can develop as a result of intrauterine hormonal imbalance of the child, perinatal injuries and compression of the skull bones in the uterine cavity. This pathology also occurs with hereditary pathologies - Apert syndrome, Crouzon syndrome and Pfeiffer syndrome. One of the leading causes of the development of craniosynostosis is known for certain - an anomaly of the gene responsible for the formation of fibroblast growth factor receptors (FGFR types I, II and III).
Pathogenetically, craniosynostosis is caused by premature synostosis of one or several cranial sutures: coronal, sagittal, lambdoid or metopic. Against this background, according to Virchow's law, compensatory growth of bone tissue occurs in the perpendicular direction, which causes deformation of the skull. Polysynostosis (and often monosynostosis) is often accompanied by intracranial hypertension, which can manifest itself as neurological disorders due to compression of the cerebral cortex, venous stagnation of the fundus, swelling of the optic disc, and in the long-term course - complete atrophy of the optic nerve and loss of vision.
Classification of craniosynostosis
Craniosynostosis, according to etiological factors, is divided into two groups:
- Syndromic. In this case, the pathology is combined with other congenital defects. These include X-linked, monogenic, chromosomal and other craniosynostoses. For example, a combination of synostosis with dysplasia of the facial bones, Smith-Lemli-Opitz syndrome or oro-finger-facial syndrome.
- Non-syndromic. This is an isolated form that occurs independently and has no associated diseases.
Depending on the number of overgrown cranial sutures, the following are distinguished:
- Monosynostosis. Characterized by damage to only 1 suture. In the case of the coronal and lambdoid suture, the overgrowth can be one- or two-sided. The most common form.
- Polysynostosis. 2-3 sutures are involved in the pathological process.
- Pansynostosis. With this form, fusion of all the bone sutures of the child’s skull is observed. It is extremely rare.
Craniosynostosis is caused by premature mineralization of cranial sutures (craniosynostosis). The most common factors for premature fusion are:
- genetic abnormalities;
- deficiency of vitamins (B8, D) and microelements (magnesium, zinc, calcium) in the diet of the expectant mother;
- mechanical impact on the fetal skull during pregnancy and childbirth;
- hormonal disorders, especially those related to thyroid hormones;
- infectious diseases of the expectant mother - herpes, rubella, influenza;
- maternal smoking during pregnancy;
- unfavorable environmental factors.
Craniosynostosis leads to deformation of the skull and increased intracranial pressure, which, in turn, leads to compression of the brain during its active growth.
Craniostenosis is the premature closure of one or all of a child’s cranial sutures
Craniostenosis is the premature closure of one or all cranial sutures, leading to abnormal growth and deformation of the skull. Currently, there are many hypotheses for the causes of craniostenosis. According to one of them, premature closure of cranial sutures may be a consequence of microcephaly. According to another, craniostenosis may be accompanied by metabolic disorders such as hyperthyroidism and rickets. Neonatologists and pediatricians consider craniostenosis as an independent congenital growth disorder of the skull, accompanied by various deformities and, in rare cases, neurological disorders. In 10-20% of cases there is a hereditary form of the disease. Craniostenosis can be a component of various hereditary syndromes accompanied by craniofacial dysostosis (Crouzon and Apert syndromes). But in most cases, the hereditary nature of the disease is not confirmed, and craniostenosis manifests itself sporadically, without a regular repetition in a number of generations.
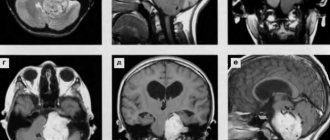
In craniostenosis, CT scan shows the absence of one or more sutures and compensatory divergence of the opposing sutures. Axial sections through the vault and base of the skull provide additional information regarding the nature of the deformity. In addition, CT scanning can confirm the involvement of the sutures of the skull base in the process of synostosis, as well as exclude concomitant pathological changes in the brain.
Diagnostics. As a rule, the main manifestation of craniostenosis is the atypical shape of the child’s head. However, a change in the shape of the head does not always indicate pathology. In many cases of so-called synostosis, positional flattening occurs. If such a suspicion exists, parents should be advised not to place the baby's head on the flattened area for 6–8 weeks and then examine the baby. If the flattening is positional, then some improvement should be observed; if it is true craniosynostosis, then there will be no change.
True craniosynostosis is confirmed by a number of examinations.
- By palpation of the bony prominences above the suture that is suspected of being involved in synostosis (exception: lambdoid synostosis).
- Apply gentle pressure with your thumbs without causing any relative movement of the bones on either side of the suture.
- Overview craniograms, where you can see:
– lack of normal transparency in the center of the seam. In some cases, the suture has a normal radiographic appearance (even on CT), possibly as a result of local formation of bone spicules;
– in case of increased intracranial pressure, local changes in the bone structure, suture diastasis and sellar erosion may be observed.
- Computed tomography, which:
– demonstrates the contours of the skull;
– shows thickening and/or folding in the area of synostosis;
– shows hydrocephalus, if present;
– may show increased frontal SA.
- MRI: usually performed in cases where there is concomitant intracranial pathology. This examination is often not nearly as informative as a CT scan.
The dimensions of the skull, such as the fronto-occipital circumference, may remain normal even in the presence of obvious deformation.
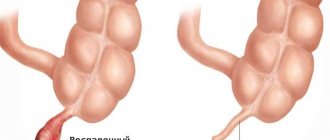
The shape of the skull depends on the location of the synostotic suture. The sagittal suture is most often affected, leading to the development of scaphocephaly . In this case, a skull is formed that is elongated in the fronto-occipital direction and compressed from the sides (Fig. 1). Significant symmetrical compensatory growth in the area of the metopic suture leads to the formation of characteristic frontal and occipital convexities. Head circumference with scaphocephaly may be higher than the age norm.
1st week
| Index | Answer rating, points | ||||
| 3 | 2 | 1 | 0 | ||
| Dynamic functions | |||||
| Relationship between sleep and wakefulness (communication skills) | Sleeps peacefully, only wakes up to feed or when wet, falls asleep quickly | Sleeps peacefully and does not wake up wet and for feeding or full and dry does not fall asleep | Doesn't wake up hungry and wet, but full and dry doesn't fall asleep or often screams for no reason | It is very difficult to wake up or sleeps little, but does not scream, or screams constantly | |
| Voice reactions | The cry is loud, clear with a short inhalation and an extended exhalation | The cry is quiet, weak, but with a short inhalation and an extended exhalation | Painful, shrill cry or isolated sobs when inhaling | There is no cry or isolated screams, or the cry is aphonic | |
| Unconditioned reflexes | All unconditioned reflexes are evoked, symmetrical | Requires longer stimulation or depletes quickly or is inconsistently asymmetrical | Not all are evoked, or after a long latent period and repeated stimulation, are quickly depleted, or are persistently asymmetrical | Most reflexes are not evoked | |
| Muscle tone | Symmetrical flexor tone overcome by passive movements | Mild asymmetry or tendency toward hypo- or hypertension that does not affect posture or movement | Constant asymmetries, hypo- or hypertension, limiting spontaneous movements | Opisthotonus or fetal or frog poses | |
| Asymmetrical cervical tonic reflex (ASTR) | When turning the head to the side, the “facial” arm inconsistently extends | — | Constant extension or lack of extension of the arm when turning the head to the side | Swordsman's Pose | |
| Chain symmetrical reflex | Absent | — | — | — | |
| Sensory reactions | Squints and worries in bright light; turns eyes towards the light source; flinches at loud noises | One of the reactions is questionable | One of the reactions of score 3 is missing or 2-3 reactions are questionable | All reactions from rating 3 are missing | |
| Risk factors | |||||
| Stigmas | None | Quantity no more than 5-6 | More than 6 and located mainly in the face area | More than 8 or the presence of gross malformations | |
| Cranial nerves | No pathology or intermittent mild convergent strabismus, or intermittent mild Graefe's sign | A combination of 2 features from score 3 or mild facial asymmetry or inconsistent horizontal nystagmus | Persistent strabismus or pronounced nystagmus, or persistent Graefe's sign, or bulbar or pseudobulbar syndrome | Combination of symptoms listed in assessment 1 | |
| Pathological movements | There are either no isolated rare athetoid movements of the fingers, or rare high-frequency tremor of the chin and hands during screaming, feeding, or passive movements. | Frequent athetoid finger movements or small, high-frequency tremors not associated with anxiety | A combination of 2 symptoms listed in score 2, or large spontaneous tremor, or single twitching of the facial muscles | Convulsions | |
Treatment
Treatment of patients with K. can be either conservative or surgical. Patients with K. with well-compensated intracranial circulation do not need treatment. If there are transient signs of impaired intracranial circulation, then appropriate medications should be used with the introduction of certain restrictions on work schedule, diet, etc. In cases of prolonged decompensation of intracranial circulation, accompanied by the threat of loss of vision, the need for surgical intervention arises. If patient K. experiences decompensation of the “venous encephalopathy” type, then surgical interventions on the bones of the cranial vault are permissible - King fragmentation (dissection of the bones of the cranial vault into separate fragments), Bagdasar-Arseni operation (resection of the bones of the cranial vault within 1-2 cm along the sagittal, coronal, parietal, lambdoid sutures), Arendt's operation (use of dissection of the frontal bone to increase the capacity of the skull), etc. In case of decompensation such as cerebral ischemia, various surgical interventions are performed on the great vessels of the head, depending on the nature of the pathology detected in them during angiography .
Forms
Depending on whether craniostenosis in a newborn is associated with other developmental anomalies, two forms are distinguished:
- non-syndromic, or isolated – occurs without extracranial deformations, has a more favorable prognosis;
- syndromic - accompanied by extracranial deformations, anomalies of the cardiovascular, respiratory, nervous systems, fusion of fingers and toes, cleft palate and upper lip and other developmental defects.
Depending on the number of prematurely mineralized seams, the following forms are distinguished:
- monosynostosis - fusion of one suture;
- polysynostosis - multiple fusion of sutures;
- pansynostosis - fusion of all cranial sutures.
There is also a classification that takes into account the deformation of the skull and which sutures have undergone early mineralization. In accordance with it, the following forms of craniostenosis are distinguished:
- scaphocephaly – affects the sagittal suture; the most common form. It is characterized by an enlargement of the skull in the anteroposterior direction and a narrowing of the head. The temples are depressed, the child's face has a scaphoid shape. Neurological symptoms are not typical for this form;
- brachycephaly - the coronoid and lambdoid sutures are fused. The skull increases in the transverse direction. This form is characterized by neurological and ophthalmological symptoms;
- trigonocephaly – affects metopic sutures. Characterized by a triangular expansion of the skull in the forehead area. This form is characterized by hypertelorism (increased distance between the eyes) and visual impairment;
- microcephaly - occurs due to multiple fusion of cranial sutures. Characterized by a uniform decrease in the size of the skull.
Craniostenosis - symptoms and treatment, photos and videos
Author: Medical News
/ 16 Aug 2020 at 19:25
Craniostenosis in children - symptoms:
- Headache
- Sleep disturbance
- Loss of appetite
- Vomit
- Memory impairment
- Developmental delay
- Skull deformation
- Tuberosity along the suture of the skull
- Premature fusion of the fontanel
- Deformation of the facial structure
Developmental delay.
– Professional medical reference books
Morritt DG, Yeh FJ, Wall SA, Richards PG, Jayamohan J, Johnson D.
Forms of craniostenosis
The forms of the disease depend on the nature of the deformation and healing of the sutures of the skull:
- Scaphocephaly. The head is elongated and narrowed. It develops when the suture that divides the skull into two halves heals.
- Brachycephaly. Increase in the transverse diameter of the skull, decrease in the height of the skull.
- Trigonocephaly. A wedge-shaped protrusion of the forehead forward is characteristic. It comes down from a large fontanel.
- Pansynostosis. The skull is reduced in size evenly. Premature fusion of sutures is characteristic.
deformation of the skull structure;
10th month
| Index | Answer rating, points | ||||
| 3 | 2 | 1 | 0 | ||
| Dynamic functions | |||||
| Communication skills | Reaction of displeasure to various situations; voice signals biological needs; play contact with an adult; imitates gestures | Play contact is selective, quickly depleted or negative emotional reactions predominate | In communication he is inactive or does not imitate gestures, or emotional reactions are poorly expressed | Game contact is weakly expressed and is not accompanied by vocal reactions and gestures | |
| Voice reactions | Imitation of sounds and syllables; variety of sound combinations; babbling words | Low-active babbling or few sound combinations, or no babbling words | Combination of symptoms listed in assessment 2 | No babble | |
| Unconditioned reflexes | Absent, except for sucking | Unconditioned reflexes are caused | |||
| Muscle tone | Full range of passive and voluntary movements | Slight increase or decrease in resistance to passive movements without affecting their volume | Limiting or increasing the range of passive movements | Opisthotonus or fetal or frog poses | |
| ASTR | Absent | — | When turning the head to the side, the extension of the “facial” arm is inconsistent | Unstable swordsman's posture | |
| Chain symmetrical reflexes | Stands independently; walks holding on with one hand | One of the reactions of score 3 is questionable | Does not stand independently or attempt to walk with support | Does not rise or a combination of signs from assessment 1 | |
| Sensorimotor behavior | Imitative hand movements - “okay”, “goodbye”; puts fingers into the hole under eye control; shows parts of another person's body; finger grip toy | Imitative movements are not expressed or does not show parts of the body or does not put fingers into holes, or there is no finger grip | Combination of symptoms listed in assessment 2 | Manipulative activity is weakly expressed | |
| Risk factors | |||||
| Stigmas | None | The number does not exceed 5-6 | More than 6 and located mainly on the face | More than 8-10 or the presence of gross malformations | |
| Cranial nerves | No pathology | Mild squinting or facial asymmetry | Persistent strabismus or Graefe's sign, or nystagmus, or ptosis, or bulbar, or pseudobulbar syndromes | Combination of symptoms listed in assessment 1 | |
| Pathological movements | None | Functional tics of the muscles of the eyes, neck, trunk, facial muscles | Tremor when manipulating objects or tongue tremor | Seizures or tremors at rest | |
Forecast
The prognosis for life with K. is usually favorable. In cases of untimely treatment due to a significant decrease in visual acuity or blindness, the ability of patients to work is sharply reduced. Considering that decompensation of intracranial circulation in K. can occur at any age under the influence of unfavorable environmental factors, patients with K. are contraindicated from working in hazardous production conditions.
Bibliography:
Bavli Ya. G. On the issue of scaphocephaly, diss., St. Petersburg, 1908, bibliogr.; Kozyrev V. A. Craniostenosis, L., 1962, bibliogr.; Romodanov A. P. and L i sch e n k o D. S. The role of vascular pathology in the development of craniostenosis, Vopr, neurokhir., No. 2, p. 28, 1969, bibliogr.; Surgery of the central nervous system, ed. V. M. Ugryumov, part 1, p. 842, L., 1969; M ii ke R. Neue Gesicht-spunkte zur Pathogenese und Therapie der Kraniosynostose, Acta neurochir. (Wien), Bd 26, S. 293, 1972; Pediatric neurosurgery, ed. by IJ Jackson a. R. K. Thompson, p. 134, Springfield, 1959; Sehrbundt Y ia 1 e E. a. T ondi M. Remarks on the surgical treatment of the craniosynosto-ses, Neurochirurgia (Stuttg.), Bd 18, S. 204, 1975; Tod PA a. Y e 1 1 and JDH Craniostenosis, Clin. Radiol., v. 22, p. 472, 1971.
A. P. Romodanov; M. X. Faizullin (rent.).
Treatment of craniostenosis
Clinically, craniosynostosis manifests itself from the moment the child is born. All forms are characterized by plagiocephaly and early closure of the large fontanel (normally this occurs at 12-18 months). Only with polysynostosis or concomitant hydrocephalus can it remain open until 3 years of age. Also, with craniosynostosis, an increase in intracranial pressure is often observed, which can be manifested by neurological disorders: anxiety, intense crying, nausea and vomiting, sleep disturbance, loss of appetite, positive Graefe's symptom, convulsions.
Each form of the disease has characteristic clinical features. Craniosynostosis of the sagittal suture (scaphocephaly or scaphoid skull) is characterized by an increase in the anteroposterior size of the child’s head with insufficient width. The elongation of the skull, “indentation” of the temporal regions, “overhanging” of the forehead and occipital part, narrowing of the face and its acquisition of an oval shape are visually determined. Palpation reveals a bone ridge above the passage of the sagittal suture. At an early age, mental development may be delayed.
Overgrowth of the lambdoid suture is most often unilateral and manifests itself as flattening of the occipital region. It is a difficult form to diagnose, since plagiocephaly is almost invisible under the hair, and neurological disorders are minimal. As the patient grows older, the dynamics of the disease are practically absent.
Coronal or coronal craniosynostosis can be either unilateral or bilateral. Overgrowth of only one half of the suture is accompanied by a typical deformation of the child’s skull - flattening of the frontal bone and the upper part of the orbit on the affected side. At the same time, the opposite half “overhangs” compensatoryly. Over time, a curvature of the nose in the opposite direction, flattening of the cheekbones, malocclusion and strabismus develop. Bilateral coronary craniosynostosis is manifested by a wide, flat and high forehead with flattened orbital margins of the frontal bone, and rarely by tower deformation of the skull (acrocephaly). Neurological disorders are nonspecific and similar to other forms.
Nontopic craniosynostosis or trigonocephaly is characterized by the development of a triangular forehead with a bony keel extending from the glabella to the greater fontanel. Hypotelorism is also observed - posterior displacement of the orbits with a decrease in the interorbital space. Over time, there is some smoothing of the bone ridge and normalization of the shape of the forehead. In half of the cases, visual impairment and mental retardation occur.
Syndromic craniosynostosis is the rarest and most severe form. In addition to plagiocephaly, there is dysplasia of the bones of the facial part of the skull, which causes respiratory failure, eating disorders and vision pathology. It is characterized by synostosis of the coronal suture and, as a result, a brachycephalic shape of the child’s head. Hypoplasia of the bones of the upper jaw, protrusion of the eyeballs from the orbits, and hypertelorism also occur. Often there is a significant expansion of the fontanel and divergence of the sagittal suture. Without treatment, children develop severe mental retardation and often die during the first 12 months of life from acute respiratory viral infections complicated by pneumonia.
The main treatment for craniosynostosis is surgical correction of the bony deformity of the skull. The optimal time for surgical intervention is the first 6-9 months of a child’s life. These periods are due to the fact that during this period the most intensive development of brain tissue is observed, which can be hampered by deformation of the cranium. In addition, the bones of the skull at this age quickly restore their structure without the development of complications. The volume and technique of the operation depend on the form of craniosynostosis and associated pathologies. At 2-3 years of age, correction is carried out solely for the purpose of eliminating a cosmetic defect. In addition to surgical treatment, the child’s diet is changed in accordance with age requirements. With the development of intercurrent diseases, drug therapy is indicated.
Forecast and prevention of craniosynostosis
The prognosis for children with craniosynostosis directly depends on the form of the disease, the timeliness of diagnosis and the effectiveness of surgical intervention. With high-quality treatment, the outcome of the disease is usually favorable. The syndromic form of craniosynostosis is considered to be unfavorable prognostically.
There is no specific prevention for this pathology. Nonspecific measures include medical and genetic consultation of the family and pregnancy planning, protecting the woman’s health while bearing a child, balanced nutrition, giving up bad habits and excluding all potential etiological factors for the development of craniosynostosis.
Craniostenosis is characterized by the following symptoms:
- deformed skull, unnatural head shape;
- increased intracranial pressure, causing persistent headaches;
- nausea and vomiting;
- exophthalmos, strabismus, congestion in the retina, which lead to decreased vision;
- sleep disorders;
- difficulty eating;
- meningeal symptoms;
- delay in psychomotor development;
- periodic convulsive seizures.
Some symptoms do not appear immediately after the birth of a child and become pronounced only in the second year of life, which complicates the diagnosis of craniostenosis in newborns.
Treatment of craniostenosis is mainly surgical. The primary goal of surgery is to relieve increased intracranial pressure and create conditions for further growth and development of the brain. In some cases, operations can eliminate a cosmetic defect in the skull and correct the shape of the head.
The optimal age for intervention is the early period – 3–9 months. After the third year of life, when the period of most active brain growth has ended, operations may be ineffective.
The following types of surgical interventions are used in the treatment of craniostenosis:
- linear craniotomy – indicated at an early age;
- circular craniotomy - more often used in older age;
- fragmentation of the cranial vault - indicated for multiple fusion of cranial sutures and only at an older age;
- flap bilateral craniotomy - indicated for severe cases of decompensated craniostenosis.
If the diagnosis is not made in a timely manner and in the absence of treatment, the following complications may develop:
- persistent deformation of the skull, leading to a significant cosmetic defect;
- persistent headaches;
- periodically recurring convulsive seizures (involuntary contractions of the muscles of the arms and legs, sometimes with loss of consciousness);
- development of visual impairment, up to its complete loss;
- mental retardation.
Classification
There are simple and complex forms: in some, a single fusion of the sutures is detected, in the second - combined. Sometimes premature fusion of all cranial sutures occurs at once.
The main classification reflects the localization of the pathological process and includes 4 points, named after the names of the cranial sutures:
- Sagittal craniostenosis, otherwise known as scaphocephaly or dolichocephaly. This is the most common type, accounting for up to 50% of all non-syndromic cases. The skull is narrowed and elongated in the anteroposterior direction.
- Coronary (coronary) craniostenosis, otherwise brachycephaly. Second most common, 20–25% of cases. The skull is wide and shortened.
- Metopic craniostenosis, otherwise known as trigonocephaly. Up to 15% of cases. The frontal bone expands, and the skull takes on a keeled triangular shape.
- Lambdoid craniostenosis, or turicephaly. The rarest variant, accounting for less than 5%.
Various combined types are also distinguished, up to the development of pansynostosis in newborns - premature fusion of all sutures of the skull at the same time. The result will be microcrania, that is, a small, rounded skull.
11th month
| Index | Answer rating, points | ||||
| 3 | 2 | 1 | 0 | ||
| Dynamic functions | |||||
| Communication skills | Braking reaction when hearing the word “no”; fulfills some requests; selective attitude towards the environment; understands the names of individual objects | Weak severity of reactions from rating 3 | There is no selective attitude towards the environment or does not imitate gestures, or an inadequate reaction to verbal communication, or does not understand the names of individual objects | Combination of symptoms listed in assessment 1 | |
| Voice reactions | Imitation of syllables and sounds; speaks babbling words “ma-ma”, “da-da”, “cha-cha” | Low-active babbling or insufficient intonation expressiveness of babbling, or does not imitate sounds and syllables | Combination of symptoms listed in assessment 2 | No babble | |
| Unconditioned reflexes | Absent, except for sucking | Unconditioned reflexes are caused | |||
| Muscle tone | Full range of passive and voluntary movements | Slight increase or decrease in resistance to passive movements without affecting their volume | Limiting or increasing the range of passive movements | Opisthotonus or fetal or frog poses | |
| ASTR | Absent | — | When turning the head to the side, the extension of the “facial” arm is inconsistent | Inconsistent fencing posture when turning the head to the side | |
| Chain symmetrical reflexes | Stands confidently without support; crouches; walks holding on with one hand; takes several steps without support | One of the reactions of score 3 is questionable | Does not stand without support or does not walk with support, or all responses from a score of 3 are questionable | Unable to stand or a combination of symptoms listed in assessment 1 | |
| Sensorimotor behavior | Throwing toys out of bed; puts fingers into holes by touch; imitative movements - turning pages, starting a car; shows parts of his body | Weak severity of 2-3 reactions from rating 3 | Imitative movements are not expressed or there is no finger grip, or does not show parts of the body | Manipulative activities and imitative movements are mild or a combination of the symptoms listed in assessment 1 | |
| Risk factors | |||||
| Stigmas | None | The number does not exceed 5-6 | More than 6 and located mainly on the face | More than 8-10 or the presence of gross malformations | |
| Cranial nerves | No pathology | Mild squinting or facial asymmetry | Persistent strabismus or Graefe's sign, or nystagmus, or ptosis, or bulbar, or pseudobulbar syndromes | Combination of symptoms listed in assessment 1 | |
| Pathological movements | None | Functional tics of the muscles of the eyes, neck, trunk, limbs, facial muscles | Tremor when manipulating objects or tremor of the tongue or elements of choreo-athetoid hyperkinesis | Seizures or tremors at rest | |
Causes and mechanism of development
There is insufficient data on the influence of heredity on the development of craniostenosis as a separate disease; a connection with genetics is detected only in 10–20% of cases. At the same time, craniostenosis can be one of the manifestations of hereditary syndromes (Cruzon, Pfeiffer, Apert and more than 180 others) with a dominant or autosomal recessive type of inheritance.
Acquired craniostenosis develops as a result of trauma and surgery. Congenital can result from fetal malformations and infections affecting the fetus. The latter include herpes, toxoplasmosis, rubella and others.
A statistical connection has been established with an increase in the level of thyroid hormone and, especially, thyrotoxicosis. Using the same method, a 60% increase in the risk of developing the disease was determined when the mother smoked.
The development of craniostenosis is based on disturbances in blood supply and metabolic processes. It has not yet been possible to study in detail the mechanism of development of this pathology.
12th month
| Index | Answer rating, points | ||||
| 3 | 2 | 1 | 0 | ||
| Dynamic functions | |||||
| Communication skills | Selective attitude towards others; communicates with adults using sound combinations; obeys some instructions; pays attention to the speaker's face | Prefers to communicate with adults using gestures or does not obey verbal instructions, or is not selective enough to relate to others | A combination of features from rating 2 or does not pay attention to the speaker's face | Lack of response to verbal communication or low interest in surroundings | |
| Voice reactions | Speaks 5-6 babbles, the intonation of a request is expressed; correlating babbling sounds with faces and objects | The babbling is inactive or says only 1-2 babbling words, or does not always accurately correlate the word with the object, action, person | No babbling syllables | Lack of babble, does not understand spoken speech | |
| Unconditioned reflexes | Absent, except for sucking | Unconditioned reflexes are caused | |||
| Muscle tone | Full range of passive and voluntary movements | Slight increase or decrease in resistance to passive movements without affecting their volume | Limiting or increasing the range of passive movements | Opisthotonus or fetal or frog poses | |
| ASTR | Absent | — | When turning the head to the side, the extension of the “facial” arm is inconsistent | Inconsistent fencing posture when turning the head to the side | |
| Chain symmetrical reflexes | Walks without support; crouches and stands up | Stands independently but walks with light support or squats but does not stand | Stands independently unsure or a combination of signs from assessment 2 | Does not stand independently or walk with support | |
| Sensorimotor behavior | Putting one item inside another; opens a box, drawer; recognition of pictures; uses a spoon and comb as intended | Weak severity of 2-3 reactions from rating 3 | Does not recognize pictures or does not use surrounding objects, or only makes patting movements with objects | Manipulative behavior is mild or unresponsive to verbal requests, or a combination of symptoms listed in rating 2 | |
| Risk factors | |||||
| Stigmas | None | The number does not exceed 5-6 | More than 6 and located mainly on the face | More than 8-10 or the presence of gross malformations | |
| Cranial nerves | No pathology | Mild squinting or facial asymmetry | Persistent strabismus or Graefe's sign, or nystagmus, or ptosis, or bulbar, or pseudobulbar syndromes | Combination of symptoms listed in assessment 1 | |
| Pathological movements | None | Functional tics of the muscles of the eyes, neck, trunk, limbs, facial muscles | Tremor when manipulating objects or tremor of the tongue or elements of choreo-athetoid hyperkinesis | Seizures or tremors at rest | |
What to do?
If craniostenosis is detected, parents should be prepared for the fact that this pathology can only be treated using surgical methods. The most gentle would be endoscopic surgery, which is performed on children who have not yet reached the age of six months. With this method, trauma will be minimal. In addition, parents should be aware that there is also a surgical treatment method. It involves artificial separation of the cranial sutures and the formation of an anatomically correct shape of the head. The scars after the operation will be hidden under the growing hairs of the baby.
Complications of craniostenosis
Surgery will help prevent complications that usually accompany craniosynostosis. If no action is taken, the shape of the baby's head will eventually change irreversibly.
As a child's brain grows, the pressure inside the skull constantly increases, which will later cause problems such as blindness and mental retardation.
Complications that may occur during surgery:
- blood loss (caused by damage to the venous sinuses)
- damage to the dura mater accompanied by brain damage
- abnormalities of bone tissue (bone destruction or inadequate regeneration)
- recurrence of craniosynostosis and the appearance of new deformities.
If the diagnosis is not made in a timely manner and in the absence of treatment, the following complications may develop:
- persistent deformation of the skull, leading to a significant cosmetic defect;
- persistent headaches;
- periodically recurring convulsive seizures (involuntary contractions of the muscles of the arms and legs, sometimes with loss of consciousness);
- development of visual impairment, up to its complete loss;
- mental retardation.
Prognosis (prospect) for craniostenosis
If left untreated, craniostenosis usually leads to disability due to vision loss, mental retardation, and other complications. An unnatural shape of the skull and a violation of the proportions of the face leads to a serious cosmetic defect that affects the child’s future life.
A successful operation allows the child's brain to develop normally. Most children who undergo surgery have a normal skull shape and do not have developmental delays or other complications.
Craniosytenosis can only be treated with surgical methods aimed at creating artificial open growth zones of the skull bones. Surgeries at the age of 3-6 months can completely eliminate the cosmetic defect and ensure normal brain development. In addition, at this age, regenerative processes are very active, and the compensatory potential is high. Therefore, early surgical treatment leads to a complete recovery of the child without any residual neurological defect.
The later the operation is performed after reaching six months of age, the more the child’s brain suffers, since it is in the first months of life that it develops especially actively, and the obstacles to its growth that exist at this time lead to irreversible consequences. Surgeries after one year of age can only eliminate cosmetic defects.
Modern neurosurgical techniques
In modern neurosurgery, many surgical techniques are used to correct craniosynostosis, the choice of which is made based on the results of the examination, taking into account the form of craniosynostosis and the required volume of intervention. This:
- linear craniotomy;
- circular craniotomy;
- unilateral and bilateral flap craniotomy;
- fronto-orbital extension;
- reconstructive operations with remodeling of the skull bones and various methods of osteosynthesis;
- total reconstruction of the skull bones;
- endoscopic cranioplasty;
- combined reconstructive techniques, for example, a combination of craniotomy and distraction devices or cranial orthoses (orthopedic devices).
Craniotomy operations involve cutting the skull bones parallel to the fused suture and removing 5-10 mm of bone tissue along the cut. Over time, the bone tissue grows and the integrity of the skull is restored. To prevent new premature fusion of bones, their edges are treated with special compounds.
In the case of polysynostosis, flap surgical techniques and multiple fragmentation of the skull bones are often used. In this case, individual small fragments of bone are removed, creating the opportunity for unhindered growth of the skull and brain. Bone flaps correctly aligned during surgery are fixed with bioabsorbable plates and suture material. By the time the physiological shape of the skull is restored, they are utilized by the body, without requiring a new intervention for removal.
If not only the brain, but also the facial skull is deformed, the frontal bone and orbital region are reconstructed and the position of the upper jaw is corrected. Titanium distraction devices of various designs are especially often used for reconstruction of the facial skull. They are installed during surgery. Spring structures begin to gradually move the bones apart immediately after surgery; other models are adjusted manually, starting from the 7th day after surgery in increments of 1 mm per day. Over the course of six months, the distractors form the correct shape and size of the skull, and then are removed.
Endoscopic operations
In the last 10-15 years, endoscopic reconstructive operations for craniostenosis have been especially widely used abroad. They are given to children up to 6 months. These are complex operations lasting about 3 hours using special pediatric neuroendoscopes, high-speed drills with specially designed burs that prevent damage to the dura mater and other endoscopic instruments. All manipulations are carried out under video control through an incision of no more than 4-5 cm. Such operations are characterized by low blood loss and a short rehabilitation period. 3-4 days after the operation and control computed tomography, the children are already discharged from the hospital.
In foreign neurosurgical clinics, operations for craniosynostosis are performed only by highly qualified neurosurgeons who specialize in working with young children. They actively use minimally invasive methods of endoscopic correction of cranial deformities, providing excellent functional and cosmetic results with minimal risk of blood loss and postoperative complications.
Craniosynostosis
Craniostenosis (craniosynostosis) is the early closure or complete absence of cranial sutures in a child, which leads to deformation of the skull, limitation of its volume and intracranial hypertension.
The skull of a newborn baby consists of six separate bones: two parietals, two temporal, occipital and frontal. Between them lie thin layers of fibrous elastic tissue - cranial sutures. The areas where several bones of the skull connect are called fontanelles or “soft spots.”
Sutures are necessary so that the skull bones can partially overlap during passage through the birth canal. This makes the birth process easier and reduces the risk of brain damage to the newborn. Closing of the fontanelles occurs during the first year of a child’s life.
In 1 out of 1000 children, intrauterine or early fusion of cranial sutures is detected - craniostenosis. Pathology is more common in boys. There are three forms:
simptomer.ru
Treatment of craniosynostosis
Typically, treatment for irregular head shape in infants involves surgery on the skull.
Remember! Only at the age of six months does an operation to change the shape of the skull make sense - in the future, changes are of a cosmetic nature. In addition to the operation, you may be offered the option of wearing a helmet for your child for 23 hours every day or physical therapy courses, changing the child’s lifestyle.
Our clinic performs operations of any degree of complexity. When you contact our Top Ichilov clinic, you will receive an answer within two business days, so even with urgent requests, you have the opportunity to come for treatment as quickly as possible. We provide all the necessary documents for the visit.