Определение болезни. Причины заболевания
Болезнь Фабри (Андерсона-Фабри) — это редкое генетически детерминированное заболевание, которое характеризуется снижением фермента альфа-галактозидазы А, который отвечает за разрушение сфинголипида глоботриаозилцерамида, накопление которого в организме приводит к изменению функционирования клеток, прогрессирующему повреждению организма, что сопровождается разнообразными клиническими симптомами. Входит в группу лизосомальных болезней накопления, сфинголипидозов.[3]
Распространенность болезни Фабри составляет от 1 на 40000 до 1 на 120000 новорожденных.[4] Разница в данных объясняется трудностями постановки диагноза и разной частотой в популяциях.
В 1898 году дерматовенеролог Джон Фабри (J. Fabri) впервые описал случай узелковой пурпуры, впоследствии осложненной макроальбуминурией у тринадцатилетнего мальчика.
Заболевание обусловлено наличием мутации в Х-хромосоме. Мутация находится в гене GLA, локализованном в Хq22.[3]
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением — это опасно для вашего здоровья!
Болезнь Фабри – дефицит фермента
Причиной болезни Фабри является снижение или отсутствие фермента альфа-галактозидазы, вызванное мутацией гена, который отвечает за его активность. При недостатке фермента в лизосомах клетки накапливаются промежуточные продукты обмена, нарушается метаболизм мембранных гликосфинголипидов.
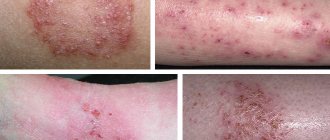
При скоплении продуктов обмена в малых и крупных сосудах в организме начинают постепенно развиваться патологические процессы. Первыми признаками являются характерные кожные высыпания пурпурного цвета, приступы лихорадки и острая боль в руках и ногах – в первую очередь страдают кисти и стопы.
Единственным видом лечения на сегодня является фермент-заместительная терапия.
Методы лечения
Самым эффективным и единственным на сегодня способом лечения является фермент-заместительная терапия. Этот метод позволяет заменить пациенту фермент, которого не хватает в организме у больного. Препараты начали успешно использовать в начале 2000 годов. Их приём позволяет значительно улучшить самочувствие пациента, однако не приводит к полному исцелению.
Фермент-заместительная терапия
Для этого используются рекомбинантные препараты альфа-галактозы. Сегодня успешно применяют Реплагал и Фабразим. Оба вводятся внутривенно, с определённой периодичностью. Процесс инвазии занимает около получаса. Препараты стоят очень дорого, поэтому лечение больных синдромом Фабри оплачивается из федерального бюджета.
Они помогают облегчить состояние больного и частично восстановить утраченные ранее функции почек, снизить болевой синдром, приостановить развитие изменений в левом желудочке сердца, предотвратить хроническую почечную и сердечную недостаточность. Улучшения в состоянии пациента наступают быстрее в случае, если лечение начало проводиться при первых симптомах заболевания.
Симптоматическое лечение
Для улучшения самочувствия больного используют препараты, способные устранить патологические проявления болезни. Для уменьшения болевого синдрома и парестезий назначают антиконвульсанты, кортикостероиды, анальгетики и нестероидные противовоспалительные препараты. Для снятия боли местно рекомендовано использование пластырей и мазей, в состав которых входит лидокаин.
При проблемах с почками и артериальной гипертензии назначается ингибиторы АПФ и блокаторы ангиотензина II. В термальной стадии периодично проводят гемодиализ или трансплантацию почки.
При повышенной свёртываемости крови для предотвращения образования тромбов и тромбоэмболии а также снижения риска инсульта назначают прием аспирина или кардиомагнила.
Ангиокератомы (пятна на коже) удаляют при помощи лазера.
Лечение
Применяют различные лекарственные препараты в плановом порядке для предотвращения кардиоэмболических событий, ишемических инсультов.
Для этого назначают антитромбоцитарные средства:
- Аспирин.
- Аспирин-дипиридамол.
- Клопедогрель.
- Тиклопидин.
- Варфарин.
Расстройства психики могут быть скорректированы назначением:
- Карбамазепина.
- Галоперидола.
- Фентанила.
- Фенитоина.
- Габапентина.
В качестве патогенетической терапии используются два ферментных препарата – Реплагаль и Фабразим. Они способствуют нормализации функции почек, сердца и цереброваскулярных тканей.
Оперативное вмешательство показано на поздних стадиях заболевания, когда необходима трансплантация почек или печени. Однако, следует иметь в виду, что трансплантация органов не может замедлить развитие БФ в других системах.
Классификация и стадии развития болезни Фабри
Выделяют две основные формы болезни Фабри: [2]
- классическую;
- атипичную.
Классическая характеризуется ранней манифестацией заболевания (в первом десятилетии жизни), наличием характерных симптомов и осложнений.
При атипичной форме происходит изолированное поражение головного мозга (ранние инсульты), сердца и почек, она манифестирует в более позднем возрасте и представляет большие трудности для диагностики.
Некоторые англоязычные авторы выделяют ещё женскую форму заболевания. Она имеет более лёгкие проявления, начало в среднем на 5-10 лет позднее классической «мужской» формы.
Болезнь Фабри: диагностика поражения у женщин
Для лиц женского пола реализуются следующие мероприятия:
- Болезнь Ормонда (ретроперитонеальный фиброз): симптомы, диагностика и лечение
- Анализ родословной.
- Анализ ДНК.
- Содержание фермента в крови не является основным показателем болезни, что обусловлено несимметричной активацией X-хромосом. Он может находиться в пределах нормы, даже если у женщины присутствует недуг.
Осложнения болезни Фабри
Со стороны нервной системы:
- ишемические инсульты в молодом возрасте, нередко являющиеся причиной смерти.
Со стороны сердечно-сосудистой системы:
- ишемические инфаркты;
- кардиомиопатии;
- аритмии;
- сердечная недостаточность, приводящая к смерти.
Со стороны почек:
- терминальная стадия почечной недостаточности, требующая трансплантации почек.
Другие системы:
- нарушение зрения; слуха, вплоть до глухоты;
- переломы костей;
- перегрев организма.
Смерть чаще всего наступает от уремии или ишемических поражений мозга и сердца на четвертом десятилетии жизни.[8]
Симптомы
Заболевание, согласно медицинским наблюдениям, впервые проявляется у взрослых и детей в пред- и пубертатный период.
Симптоматика недуга сводится к следующему:
- Жжение и болезненность, возникающие в конечностях, усиливающиеся при физических нагрузках и контакте с горячим. Боль нередко сопровождается лихорадочными состояниями.
- Слабость. Очень часто подобные проявления сопровождаются плохим настроением, тревожностью, чувством беспокойства. Пациенты чувствуют ухудшение настроения и неуверенность в собственных силах, что в значительной мере снижает качество жизни.
- Снижение потоотделения.
- Утомляемость ног и рук. Человек, по мере развития недуга, не может выполнять даже самые обыкновенные действия, так как испытывает постоянную усталость. Вкупе с болевыми ощущениями такие симптомы приводят к полной беспомощности.
- Протеинурия. Недуг, сопровождается повышенным выведением белка вместе с мочой. Нередко такой симптом становится причиной неправильной постановки диагноза.
- Образование высыпаний в области ягодиц, паха, губ, на пальцах.
- Вегетативные нарушения.
Приблизительно у трети детей одновременно с перечисленными симптомами развивается суставной синдром. Ребенок испытывает нарастающие мышечные боли, снижение зрения, недостаток сердечно-сосудистой деятельности. По мере прогрессирования ферментной недостаточности наблюдается поражение почек, повышение артериального давления. Тем не менее детям такой диагноз ставится очень редко, потому так важно при любых проявлениях обращаться в профессиональные центры для исследования крови на генетическом уровне.
Симптомы вызванные болезнью
Болезнь Андерсона появляется обычно в детском возрасте от 1 года. Взрослые люди болеют в редких случаях. Список возможных симптомов:
- раздражение на кожном покрове рук, ног, паховой области и лице, которое появляется в виде язвочек, при контакте с горячими предметами область поражений увеличивается;
- прекращение работы потовых желез;
- сильная слабость после легких физических нагрузок;
- лихорадка, повышение температуры;
- появление ревматического синдрома суставов;
- преимущественно у детей появление вегетативных нарушений;
- боли в мышцах по мере прогрессирования заболевания;
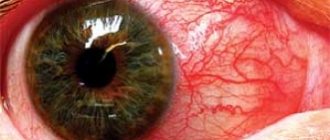
- упадок зрения, может развиться катаракта, разрушаются сосуды сетчатки глаза;
- сердечные боли;
- проблемы с почками;
- болезненные ощущения при сгибании и разгибании конечностей, может длиться несколько часов или неделями;
- после плотного обеда и ужина заболевает желудок или кишечник;
- тошнота;
- рвота;
- диарея.
К симптомам также относят внешние проявления болезни. Обычно внешние изменения заметны в возрасте 13 лет. Типичные характеристики у мальчиков:
- западение переносицы;
- выступление бровей и лба вперед;
- в 2 раза увеличенная нижняя челюсть;
- в 2 раза увеличивается рот;
- расширяются губы.
Такие изменения вызывает соматотропный гормон.
Болезнь сопровождается болью, которая подразделяется на 2 типа:
- Боль, возникающая как на физический раздражитель, называемая нейропатической. При любом прикосновении к ступням и ногам, больной чувствует жжение и покалывания.
- Внезапные боли. Появляются внезапно в виде приступов. Зачастую болевой порок возникает на ступнях и ногах, иногда в других частях тела.
Диагностика болезни Фабри
Ранняя диагностика очень важна для правильного и своевременного назначения лечения пораженных болезнью органов, предотвращения осложнений.[5]
Важным методом диагностики болезни Фабри является оценка генеалогического анамнеза пациента, так как при этом могут обнаружиться родственники со сходными симптомами, которые не знают о своем заболевании. Ген, ответственный за болезнь Андерсона-Фабри, может передаваться через большое число поколений, поэтому больными оказываются многие ближние и дальние родственники. Для определения риска наследования этого заболевания необходимо собрать информацию о здоровье всех известных членов семьи.[3]
Болезнь Фабри бывает очень трудно отличить от более распространенных заболеваний, и пациенты в течение долгих лет могут оставаться без верного диагноза.
Предварительный диагноз ставится по результатам опроса, жалоб пациента, генеалогическому анамнезу.
Для верификации диагноза используют методы обнаружения субстратов и энзимов, ДНК-диагностику.
- Измерение активности альфа-галактозидазы А. [2] При болезни Андерсона-Фабри активность этого фермента у представителей мужского пола всегда ниже нормы, а у женщин этот показатель бывает в пределах нормы или слегка снижен. Материалом для проведения исследования могут служить лейкоциты, плазма крови, слезная жидкость, культура фибробластов.
- Наиболее точным методом диагностики является секвенирование гена GLA .[2] На сегодняшний день описано более пятисот мутаций этого гена, приводящих к болезни Фабри. Использование данной методики ограничено её стоимостью. ДНК-диагностику целесообразно применять для женщин, т. к. у них определение активности альфа-галактозидазы А не всегда дает достоверный результат, и для родственников больного, т. к. они могут являться носителями мутантного гена или больны атипичной формой заболевания. Проведение данного анализа возможно в Центре молекулярной генетики в Москве.
- Количественное определение глоботриаозилцерамида. Этот метод зарекомендовал себя для наблюдения за состоянием пациентов и оценки эффективности лечения, для определения формы заболевания (типичная, атипичная). Материалом для исследования могут быть плазма крови или сухие пятна крови.
- В некоторых случаях проводится биопсия почки с целью обнаружения клеток, содержащих лизосомы с характерным субстратом.
Дифференциальная диагностика болезни Фабри проводится с наследственной геморрагической телеангиэктазией, ревматической лихорадкой, болезнью Фордайса, Шиндлера, другими наследственными болезнями накопления.[1]
Диагностика
Диагностика болезни Фабри осуществляется на основании клинической картины, а также результатов лабораторных и инструментальных исследований. При оценке симптомов патологии у мужчин во внимание принимается сочетание таких признаков, как:
- ангиокератомы в нижней части туловища;
- хроническая невропатическая боль в конечностях с периодами обострений (кризов);
- помутнение роговицы;
- периодическое повышение температуры тела до фебрильных показателей.
Для лабораторных анализов могут использоваться образцы крови (в том числе сухие пятна), мочи, слезной жидкости, тканей. Основные исследования:
- Определение активности альфа-галактозидазы в лейкоцитах или фибробластах кожи с помощью флоуметрии. У мужчин с болезнью Фабри активность фермента снижена, у женщин находится близко к нижней границе нормы.
- Исследование биоптата почки. В клетках выявляются лизосомы с характерным субстратом.
- Определение количества сфинголипидов в крови. При болезни их число повышено. Тест полезен не только для постановки диагноза, но и для мониторинга состояния пациента.
- Секвенирование ДНК гена GLA – наиболее точный метод выявления патологии. Его недостаток – высокая цена.
При обнаружении генетической мутации целесообразно проведение ДНК-диагностики у всех родственников пациента, которые могут иметь такую же Х-хромосому.
Выявление болезни Фабри у ребенка в период эмбрионального развития возможно путем определения активности альфа-галактозидазы в клетках, полученных из амниотической жидкости или ворсинок хориона (оболочки плода). Провести такую диагностику можно только инвазивными способами: путем амниоцентеза, хорионбиопсии или кардиоцентеза.
Инструментальные методы, применяемые во время обследования при подозрении на болезнь Фабри:
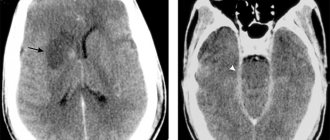
- МРТ головного мозга – демонстрирует гиперинтенсивный сигнал во фронтальных и теменных долях (в белом веществе), поражение заднего бугорка таламуса, сосудистые мальформации;
- ЭКГ – показывает увеличение (гипертрофию) левого желудочка, уменьшение интервала P – R на ранних этапах и предсердно-желудочковую блокаду на терминальной стадии.
Болезнь Фабри дифференцируют от наследственной геморрагической телеангиэктазии (синдрома Рандю – Ослера – Вебера) и ревматизма.
Особенности заболевания
Какие есть особенности болезни Фабри у мужчин?
- В связи с тем, что в организме парней присутствует одна Х-хромосома, то вероятность развития болезни выше, чем у женщин.
- Существует закономерность, что мужчина, организм которого поражён данным заболеванием, не может передать его своему сыну. Однако неправильный ген передаётся дочерям больного.
Особенности заболевания Фабри у женщин:
- У женщин в организме присутствуют две Х-хромосомы. Поэтому заболевание может проходить в более лёгкой форме и его признаки проявляются позже.
- Женщина может передать неправильный ген как дочерям, так и сыновьям. Вероятность того, что ребёнку достанется данное заболевание по наследству, составляет 50 %.
Причины
В основе заболевания лежит генетический дефект половой Х-хромосомы. В одном из участков этой хромосомы закодирована информация о ферменте α-галактозидазе А. Если возникает мутация, то это приводит к снижению количества этого фермента или уменьшению его активности.
Дефект имеет рецессивный характер наследования. Что это означает? Поскольку у мужского пола только одна Х-хромосома (а вторая – У), то у мальчиков при наличии патологической Х-хромосомы всегда развивается классическая картина заболевания. Больной мужчина обязательно передаст мутантную Х-хромосому всем своим дочерям без исключения (то есть в 100% случаев), а вот сыновья у него будут здоровыми.
У женщин две Х-хромосомы. Если одна из них мутантная, то у таких женщин возникают клинические проявления, но они менее выражены, позже развиваются и медленнее прогрессируют, почти всегда это атипичные формы болезни. Если же у женщины совпадут обе мутантные Х-хромосомы, одна от отца, другая – от матери (вероятность чего практически равна нулю, учитывая распространенность заболевания), тогда также развивается классическая картина болезни Фабри. Женщина может передать мутантную Х-хромосому как сыновьям, так и дочерям (при наличии одной мутантной Х-хромосомы вероятность составляет 50%).
Лечение болезни Фабри
Лечение болезни Фабри состоит в замещении дефицитного фермента с помощью ферментозаместительной терапии. Она проводится с помощью внутривенного вливания препарата. Обычно ферментозаместительная терапия используется вместе с различными методами лечения конкретных симптомов.[5]
В России сегодня используются два препарата для ферментозаместительной терапии: Фабразим, Джензайм; Реплагал, Шайер. Эти препараты имеют очень большую стоимость, лечение больных оплачивается из средств федерального бюджета. Так как заболевание относится к орфанным (редким), существует закон об ускоренной процедуре исследования лекарственных препаратов, предназначенных для лечения таких болезней.[3]
Мужчинам начало ферментозаместительного лечения рекомендуется в ближайшее время после установления диагноза.[5]
Для купирования боли применяются препараты из групп атиконвульсантов, антидепрессантов, анальгетиков, нестероидных противовоспалительных препаратов, наркотических анальгетиков.[2]
Больным показано применение антигипертензивных препаратов.
При развитии терминальной почечной недостаточности проводят трансплантацию почки. Донорами не рекомендуется быть женщинам, которые являются родственницами больного.[2]
В качестве коррекции тугоухости показано использование слуховых аппаратов.
Физическая активность пациентов с болезнью Фабри должна быть ограничена из-за возможного усиления симптомов и перегрева организма.[3]
Ситуации, при которых нецелесообразно назначать ферментозаместительную терапию:
- период беременности и лактации;
- если имеется другое опасное для жизни заболевание, при котором прогноз от применения заместительной терапии не станет лучше;
- есть серьезные осложнения (ишемический инсульт, реанимационные больные).
Прогноз
Болезнь Фабри имеет относительно благоприятный прогноз при:
- своевременном назначении ферментозаместительной терапии;
- регулярном мониторинге состояния пациента (рекомендуется прохождение комплексного медицинского осмотра не реже 1 раза в год);
- соблюдении всех рекомендаций врача.
Женщины с данным заболеванием способны к деторождению. Однако исследования показывают, что проявления патологии могут спровоцировать ряд осложнений как в период вынашивания ребенка, так и сразу после родов.
Без адекватного лечения болезнь Фабри отрицательно отражается на качестве жизни пациентов: они страдают от болевого синдрома, проблем с сердцем и почками, а также от снижения трудоспособности. Патологические симптомы негативно влияют на психоэмоциональное состояние больных, у них развивается депрессия.
Основные причины смерти пациентов с болезнью Фабри:
- почечная недостаточность;
- патологии сердца;
- инсульт.
Средняя продолжительность жизни: для мужчин – 50 лет, для женщин легкой формой заболевания – 50 лет.
Болезнь Фабри. Лечение и профилактика
Пациенту сначала назначаются болеутоляющие средства. Они облегчают состояние больного. Другим направлением лечения является восполнение в организме фермента, который не вырабатывается из-за испорченного гена. Называется оно ферментно-заместительная терапия.
- Мочекаменная болезнь: причины, особенности протекания, диагностика и способы лечения болезни
Существует мнение, что если диагностировать заболевание на ранней стадии и применить пациенту данное лечение, то можно одолеть болезнь Фабри. Профилактика данного недуга отсутствует, так как оно передается по наследству. Единственным способом избежать рождения ребенка с этим заболеванием является диагностирование культивируемых амниотических клеток активности a-галактозидазы А.
Самым лучшим вариантом для носителей этой болезни является его раннее диагностирование. Для этого не нужно пускать на самотек жалобы ребенка о присутствии болевых ощущений в кистях рук или ног. Особенно если известны случаи, что в роду кто-то из родственников страдал от данного недуга. Современная медицина позволяет оказать помощь людям, у которых присутствует болезнь Фабри в организме. Поэтому лучше обратиться к специалисту, сделать обследование организма и начать своевременное лечение.
Болезнь Фабри – что это такое?
Лизосомные болезни накопления – это общее название одной большой группы редких наследственных недугов, которые вызываются нарушением функции лизосом. Как раз к этой категории и относится болезнь Фабри. Связана она со снижением активности α-галактозидазы – фермента лизосом, который ответственен за расщепление гликосфинголипидов. Как следствие – жиры в избыточном количестве скапливаются в клетках и мешают их нормальному функционированию. Как правило, страдать приходится эндотелиальным или гладкомышечным клеткам сосудов, почек, сердца, ЦНС, роговицы.
Болезнь Фабри – тип наследования
Данный недуг считается генетически детерминированным с Х-сцепленным типом наследования. То есть, болезнь Фабри передается только по Х-хромосомам. У женщин таковых две, и потому аномалия может быть унаследована как сыном, так и дочерью. Вероятность рождения ребенка с генетическим отклонением в таком случае составляет 50%. У мужчин всего по одной Х-хромосоме, и если она является мутировавшей, то болезнь Андерсона Фабри будет диагностирована у их дочерей с вероятностью 100%.
Болезнь Фабри – причины
Это генетическое заболевание, а потому главной причиной его появления являются мутационные изменения в GLA-генах – отвечающих за кодирование фермента. Согласно статистике и результатам многочисленных медицинских исследований, лизосомная болезнь накопления Фабри в 95% случаев имеет наследственную природу, но есть и исключения. 5% пациентов «заработали» диагноз на начальных этапах формирования эмбриона. Виной тому стали случайные мутации.
Прогноз. Профилактика
При надлежащем лечении и вовремя начатой заместительной ферментативной терапии прогноз для больных благоприятный. При отсутствии лечения смерть от сердечно-сосудистых, неврологических и почечных осложнений наступает на четвертом десятилетии жизни.[4]
Профилактика заболевания заключается в пренатальной или предимплантационной (при ЭКО) диагностике наличия мутантного гена методом ДНК-диагностики и прерывания беременности по медицинским показаниям.[4] Необходимо обследование всех потенциальных носителей гена для своевременного назначения заместительной ферментативной терапии и предотвращения клинических проявлений болезни и ее осложнений. Возможно скрининговое исследование сухих пятен крови с определением активности альфа-галактозидазы, но оно ограничено финансовыми возможностями региона. Необходимо, чтобы все пациенты с признаками болезни Фабри были исследованы на это заболевание. Для этого существует программа, регламентирующая забор и отправку материала в Центр молекулярной генетики, обеспечение больных препаратами заместительной терапии.
Диагностика недуга у мужчин
Для лиц мужского пола проводятся следующие мероприятия:
- Анализ родословной, генеалогического древа. Учитывая наследственный характер поражения, основное значение в диагностике имеет сбор семейного анамнеза. Но у членов одной семьи заболевание может остаться нераспознанным.
- Исследование на содержание фермента галактозидаза в крови.
- Анализ ДНК – проводится в случае получения неоднозначных результатов биохимических исследований. ДНК может быть взята из любого биологического материала.
Список литературы
- Козлова С.И., Демикова Н.С. Наследственные синдромы и медико-генетическое консультирование. М: Т-во научных изданий КМК; Авторская академия, 2007. 448 с.
- Федеральные клинические рекомендации по диагностике и лечению болезни Фабри. Москва, 2013.
- Информация о болезни Фабри (дата обращения 25.01.18г.)
- Mehta A, Ricci R, Widmer U, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. European Journal of Clinical Investigation 34 (3): 236–42
- Союз пациентов, страдающих болезнью Фабри ((дата обращения 25.01.18г)
- Omid Motabar, Ellen Sidransky, Ehud Goldin, and Wei Zheng. Fabry Disease – Current Treatment and New Drug Development. Curr Chem Genomics. 2010; 4: 50–56
- Stephen Waldek, Sandro Feriozzi. Fabry nephropathy: a review – how can we optimize the management of Fabry nephropathy? BMC Nephrol. 2014; 15: 72
- Frank Weidemann, Maria D Sanchez-Niño et al. Fibrosis: a key feature of Fabry disease with potential therapeutic implications. Orphanet J Rare Dis. 2013; 8: 116
- Juan M. Politei, Didier Bouhassira et al. Pain in Fabry Disease: Practical Recommendations for Diagnosis and Treatment. CNS Neurosci Ther. 2020 Jul; 22(7): 568–576
Диагностика заболевания
Корректная постановка диагноза часто вызывает затруднения, так как недуг встречается достаточно редко и проявляется целой гаммой клинических симптомов. Пациенты с подозрением на болезнь Фабри проходят обследование у специалистов разных клинических областей.
Практический опыт показывает, что между начальными проявлениями симптомов и окончательным диагнозом в среднем проходит около 12 лет. Очень важно установить болезнь как можно раньше, чтобы начать полноценное лечение.
Медицинские работники должны заподозрить наличие этого генетического поражения, если у человека выявлена комбинация более двух симптомов заболевания. В раннем детском возрасте болезнь Фабри (фото пораженной хромосомы представлено ниже), практически не проявляется, поэтому диагностика может быть осложнена.
Осложнения
При дальнейшем прогрессировании болезни Фабри могут возникнуть следующие осложнения:
- Мочекаменная болезнь симптомы и лечение у мужчин
- Дестабилизируется работа почек. Из организма начинает выводиться белок. Развивается почечная недостаточность.
- Поражение сердечной системы. Может измениться его форма, возникнуть аритмия.
- Недостаточное питание головного мозга, что повышает вероятность возникновения инсульта.
Как проявляется?
Особенностью данного заболевания является то, что оно имеет разную симптоматику у разных людей. У некоторых на протяжении всей жизни признаки носят слабый характер. А у кого-то проявляются в тяжёлых формах. Как говорилось выше, у представителей сильного пола признаки недуга можно заметить раньше, чем у девочек.
Статистика говорит о том, что диагноз «болезнь Фабри» ставится людям в возрастном промежутке от 30 до 45 лет. Хотя признаки заболевания были у человека раньше.
На это следует обратить внимание, если существует вероятность передачи данного заболевания по наследству.
Используемые источники:
- https://probolezny.ru/bolezn-fabri/
- https://onevrologii.ru/nasledstvennye-zabolevaniya/bolezn-fabri
- https://fb.ru/article/177927/bolezn-fabri-simptomyi-lechenie-foto
- https://www.syl.ru/article/292490/bolezn-fabri-prichinyi-simptomyi-lechenie
Болезнь Фабри – симптомы
Признаки болезни в разных организмах проявляются по-своему:
- Мужчины.
У представителей сильного пола болезнь Андерсона-Фабри, как правило, начинает проявляться с детства. Первые признаки: боль и жжение в конечностях. Некоторые пациенты жалуются на появление багровой сыпи, которая в большинстве случаев покрывает область от пупка до колен. Поскольку заболевание прогрессирует медленно, серьезные симптомы – дискомфортные ощущения в животе, звон в ушах, частые позывы к испражнению, боли в спине и суставах – становятся различимы только к 35 – 40 годам. - Женщины.
В женском организме заболевание показывает широкий спектр клинических проявлений. В то время как одни пациентки и не догадываются о своей проблеме, другие мучаются с дистрофией роговицы, усталостью, сердечно-сосудистыми нарушениями, ангидрозом, желудочно-кишечными расстройствами, болезнями почек, поражениями глаз, неврологическими расстройствами. - Дети.
Хоть в большинстве случаев первые симптомы недуга появляются рано, болезнь Фабри у детей часто остается незамеченной и развивается до сознательного возраста. Самыми ранними признаками считаются боль и ангиокератомы, которые часто располагаются за ушами и упускаются из виду специалистов. Другие проявления заболевания у маленьких пациентов: тошнота с рвотой, головокружение, головные боли, лихорадка.