Причины возникновения
Генетическое изменение гена, отвечающего за процесс биосинтеза коллагена – это основная предпосылка к развитию синдрома Альпорта. Такую наследственную мутацию ребёнок приобретает от родителей: дочь – от отца, а от матери – как мальчик, так и девочка. У человека может быть поврежден ген, при этом болезнь развивается не всегда, но будет наследоваться детьми.
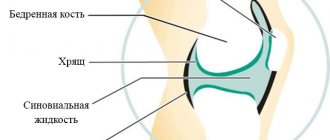
Изменению подвергается один из трех генов, отвечающих за процесс биологического синтеза коллагена типа IV. Именно он входит в структуру базальных мембран, расположенных в почках, органах зрения и слуха. Базальные мембраны являются особыми образованиями – тоненькими границами, которыми некоторые ткани организма человека отделяются друг от друга, поддерживают и укрепляют их структуру. Если их состав и целостность нарушаются, возникают опасные явления:
- происходит неполная и некачественная фильтрация токсических и других веществ, поступающих из крови;
- изменяется состав мочи, в которой наблюдается значительное увеличение эритроцитов (гематурия или следы крови в моче) и нефильтрованного белка (протеинурия).
Такие изменения приводят к развитию почечной недостаточности тяжелого типа, в некоторых случаях к полному прекращению функций органа и летальному исходу.
Синдром Альпорта и его развитие могут спровоцировать некоторые факторы:
- тяжелые заболевания, вызванные бактериями или вирусами;
- воздействие на организм компонентов вакцин;
- чрезмерные нагрузки физического характера.
Медицинская статистика обладает данными о том, что каждый пятый ребенок с подтвержденным диагнозом наследственный нефрит, появляется у родителей, не имеющих патологий, связанных с работой почек. Причина возникновения синдрома Альпорта в таких случаях связана с генными мутациями спонтанного типа.
Истинные причины заболевания
Истинные причины синдрома альпорта до сих полностью не изучены учеными. В нашем организме есть ген, функциональной обязанностью которого является обмен белка в тканях почек. Так вот мутация этого гена и является наиболее вероятной причиной появления недуга.
Теперь рассмотрим провоцирующие факторы, которые могут способствовать появлению заболевания. К ним можно отнести:
- тяжелые инфекционные процессы;
- прививки;
- сильные физические нагрузки.
Как видно из многочисленных случаев медицинской практики, порой развитию синдрома альпорта может способствовать обычная острая респираторная вирусная инфекция. Именно ввиду таких высоких рисков заболеваемости дети, которые имеют отягощенную наследственность, должны чаще проходить регулярное диагностическое обследование.
Интересным является тот факт, что синдром альпорта имеет доминантное наследование. Что это значит? Это говорит о том, что, если мужчина является носителем мутировавшего гена, то его дочери будут болеть, а сыновья родятся здоровыми. В то же время дочери передадут болезнь всем своим детям.
Впервые это заболевание было зарегистрировано в начале прошлого столетия. Врач наблюдал за семьей, в которой в течение нескольких поколений наблюдалась гематурия. Позже была замечена связь между гематурией и тугоухостью, а также поражением глаз. Позже, когда медицина совершенствовалась, медиками глубже исследовали генетическую природу данного синдрома.
В большинстве случаев у ее «обладателей» имеются родственники с почечными патологиями и другими признаками данного синдрома. Роль играют также родственные браки, в результате которых у ребёнка возрастает вероятность получения одинаковых генов. У больных с синдромом Альпорта выявляются сдвиги со стороны иммунной системы.
Методы терапии
Лечение синдрома Альпорта включает в себя диету, прием лекарственных средств, своевременную санацию очагов инфекции.
Прививки детям противопоказаны, вакцинация возможна только при наличии строгих показаний.
На данный момент не существует фармакологических препаратов, которые бы воздействовали на генетический дефект.
Широко применяют метаболические препараты, которые позволяют повысить . К ним относятся кокарбоксилаза, витамины А, Е, В6. При появлении белка в моче назначаются нефропротекторы (препараты, защищающие почки).
- Синдром раздраженного мочевого пузыря у женщин, мужчин и детей
К ним относятся ингибиторы ангиотензинпревращающего фермента (эналаприл, лизиноприл, рамиприл, пириндоприл) и блокаторы ангиотензиновых рецепторов (лозартан).
Вышеперечисленные препараты относятся к группе гипотензивных средств.
Даже при пониженном артериальном давлении у детей, следует принимать минимальные дозы лекарств, чтобы снизить скорость прогрессии недостаточности почек.
Физическая активность
Ребенок с синдромом Альпорта должен воздерживаться от тяжелых физических нагрузок. Однако ему необходимы ежедневные пешие прогулки не менее 40 минут.
Это позволит улучшить микроциркуляцию в почках, а также будет способствовать нормальному развитию.
Диетические предписания
Следует исключить из рациона:
- соленую, жирную и копченую пищу;
- рряности и острую еду;
- рродукты с высоким содержанием искусственных красителей.
Необходимо следить за количеством поступления белка в организм, при развитии почечной недостаточности жидкость ограничить до одного литра, соль до одного грамма в сутки.
С пищей должно поступать достаточное количество калорий, витаминов, макро- и микроэлементов.
Хирургическое вмешательство
На , при которых почки не справляются с выделением токсических продуктов обмена веществ, проводится и пересадка почки.
- Как лечить двухсторонний хронический пиелонефрит
Гемодиализ выполняется аппаратом « » почка. Суть процедуры заключается в очищении крови пациента от токсических веществ, является жизненно необходимой.
Также пациенту выполняется пересадка почки, после которой назначается иммуносупрессивная терапия для профилактики отторжения трансплантата.
Народная медицина
Применяется совместно с традиционными способами после консультации с лечащим врачом. Используются свойства лекарственных растений, помогающие облегчить клинические проявления заболевания.
Для улучшения микроциркуляции в почках можно употреблять неконцентрированные и .
Увеличить скорость клубочковой фильтрации поможет , плоды можжевельника, почки березы.
Прогноз и профилактика
Неблагоприятный прогноз наиболее вероятен для ребенка мужского пола, а также при наличии:
- высокой концентрации белка в общем анализе мочи;
- раннего развития почечных нарушений у близких родственников;
- тугоухости.
Если выявлена изолированная гематурия без сопутствующей протеинурии и снижения слуха прогноз заболевания благоприятный, функциональная недостаточность почек развивается редко.
К профилактическим мерам относится планирование беременности (санация хронических очагов инфекции, женщина должна избегать чрезмерных физических и эмоциональных нагрузок, своевременно стать на учет в женскую консультацию, при наличии показаний пройти медико-генетическое консультирование).
Ребенку необходимо проходить регулярные профилактические осмотры у педиатра. При обнаружении первых признаков болезни, родители должны сообщить врачу.
Синдром Альпорта – тяжелое генетически обусловленное заболевание почек, зрительного и слухового аппарата. При своевременной постановке диагноза и соблюдению рекомендаций, можно замедлить темпы развития почечной недостаточности, тугоухости.
Синдром Альпорта (нефрит наследственный) встречается редко. Эта патология провоцирует нарушение зрения и потерю слуха. Нередко это ведет к потребности в пересадке почки.
Этот синдром проявляется в 3–5 лет. Наследственный нефрит у детей постоянно прогрессирует. Параллельно ребенок теряет слух и зрение, поражается клубочковый аппарат почек.
Развивается синдром Альпорта из-за мутации гена, вырабатывающего коллаген. Эта разновидность коллагена участвует в построении капсулы хрусталика и части внутреннего уха. Отсюда нарушения их функции, а также нарушаются функции самих почек.
Согласно международной классификации эта патология относится к врожденным аномалиям, хромосомным нарушениям и деформациям. Она считается врожденным пороком, так как страдает сразу несколько органов и систем.
Диагностика
Поставить диагноз наследственного нефрита помогает проведение нескольких исследований, сбор и анализ анамнеза. Именно генетический характер болезни предполагает изучение особенностей здоровья родителей, бабушек и дедушек малыша. Необходима оценка показателей, наличие трех из них позволяет диагностировать синдром:
- признаки гематурии;
- патологии почек, почечная недостаточность, в том числе закончившаяся смертью;
- заболевания органов зрения и слуха врожденного характера;
- прогрессирующее ухудшение качества зрительного и слухового восприятия у ребенка;
- наличие изменений строения базальной мембраны (проводится при биопсии почечной ткани).
Для оценки состояния пациента и особенностей развития патологического процесса врач назначает исследования:
- анализ на синдром Альпорта (предполагает изучение крови и мочи);
- ультразвуковое исследование почек, надпочечников;
- проведение рентгенографии органа;
- биопсия тканей;
- ДНК-тест для определения носителя гена-мутанта.
Пациенту требуются дополнительные посещения врачей узкой специализации – нефролога, генетика, окулиста, отоларинголога.
- Удвоение члс левой почки что это такое
Причины возникновения наследственного заболевания у детей
Синдром Альпорта также называется наследственным воспалением почек. Встречается как у мальчиков, так и у девочек. Выявляется патология при прохождении профилактических осмотров.
Заболевание обусловлено генетическим дефектом структуры белка. К провоцирующим факторам, которые приводят к мутации гена, относятся:
- Перенесенное инфекционное заболевание матери в период беременности. Особенно опасна инфекция в первом триместре, когда происходит закладка органов и тканей плода.
- Вакцинация, проведенная беременной женщине.
- Чрезмерная физическая нагрузка и эмоциональный стресс, постоянно сопровождающие будущую маму.
Причины и факторы развития
Молекула ДНК — длинная цепочка из химических соединений, лежащая в основе любого гена. Человек на заре жизни получает в наследство множество различных генов. Они рассортированы на сорок шесть хромосом. Две из них определяют пол — Х и Y. Причина синдрома Альпорта — неправильный ген, в котором содержится информация о структуре коллагена 4-го типа. Существует три копии этого гена, расположенные во второй и Х-хромосоме.
Тайна ДНК — видео
Неправильное строение гена приводит к тому, что белок коллаген также имеет дефектную конфигурацию. В первую очередь это приводит к изменению сосудистых клубочков. Соединительная ткань почечного фильтра со временем воспаляется, формируется наследственный нефрит. С возрастом клубочки гибнут и замещаются рубцовой соединительной тканью. Таким образом, существенно страдает работа почек по очистке крови от вредных веществ и других токсичных продуктов обмена, формируется хроническая почечная недостаточность. Воспалённый почечный фильтр начинает пропускать через себя клетки крови эритроциты, которые придают моче красноватый оттенок (гематурия). Ещё одним признаком наследственного нефрита является высокий уровень артериального давления. Это явление обусловлено избытком особого вещества — ангиотензина, который выбрасывается почками в кровь.
При наследственном нефрите повреждается почечный фильтр
Аномальный коллаген 4-го типа приводит к тому, что начинает страдать орган слуха. В первую очередь нарушается передача звука по системе лабиринта внутреннего уха. С течением времени соединительной тканью замещаются нервные окончания, ответственные за выработку электрического сигнала. В конечном итоге эти изменения приводят к глухоте.
Хрусталик, окружённый подобным коллагеном, часто мутнеет с формированием катаракты. Свет через такую изменённую линзу не может проникнуть дальше и попасть на пигментную оболочку глаза (сетчатку). Изображение на ней формируется нечётким и размытым. Кроме того, из-за наследственной болезни страдает сама сетчатка глаза, которая перестаёт воспринимать свет. Из-за этого в поле зрения появляются чёрные пятна, которые соответствуют поражённым участкам.
Хрусталик — основной компонент оптической системы глаза
Симптомы
В три года и раньше у ребенка появляются первые симптомы, выраженные изолированным мочевым синдромом, то есть гематурией (изменением количества или качества мочи, её осадка). Чаще всего наследственный нефрит у детей выявляется во время обследования перед поступлением в детские сады или при ОРВИ, то есть случайно. Семейный нефрит отличается от приобретенного отсутствием временного промежутка между началом действия раздражителя и ответной реакции.
Дети не чувствуют дискомфорта на начальной стадии хвори. Главным показателем болезни является измененный цвет мочи, смена пигмента всегда наблюдается на первых этапах. Усиленная пигментация в красные тона провоцируется прививками, фитнес нагрузкой, заболеваниями гортани, легких. На начальных этапах превышения выделения белка с мочой непостоянны, всё же протеинурия возрастает с течением болезни. Возможно наличие лейкоцитурии — признак деструктивных изменений в организме.
Следующим этапом идет ухудшение самочувствия пациента: отравление организма, слабость, пониженное давление, развитие глухоты, возможны проблемы с глазами. Понижение чувствительности слухового аппарата на начальных этапах выявляется только при помощи исследования слухового аппарата. Самым распространенным возрастом начала проявления глухоты является промежуток с шести до десяти лет. Тугоухость может являться первым симптомом заболевания, может проявиться даже раньше мочевого синдрома.
Зрение при наследственном нефрите портится с вероятностью двадцати процентов. Порча зрения выражается одним из вариантов, таких как: миопия, катаракта, изменение физического устройства хрусталика.
В подростковом возрасте явным симптомом является повышенное давление, физическая недоразвитость. Характерной стигмой болезни является смешение таких аномалий, как: деформация прикуса, неправильная форма ушей, аномальная кривизна мизинца руки, большой промежуток между первым и вторым пальцем стопы. Синдром Альпорта, приобретенный наследственным путем, характеризуется схожими отклонениями у родственников.
Если мы говорим про то, как пошагово происходят нарушения в структуре работы почек, то выделяется минимум три этапа. Первый: дестабилизация частичной функциональной работы почек. Второй: деструктивная работа как центральных, так и срединных частей почек. Третий: Ухудшение фильтрации клубочкового аппарата.
Подытоживая симптомы, приходим к выводу о том, что заболевание проходит «поступенчато», то есть поэтапно. Изначально мы имеем дело со скрытой бессимптомной болезнью, а потом она перетекает в открытое заболевание с явной симптоматикой.
Необходимое лечение
Лечение синдрома Альпорта заключается в нормализации работы почек, так как особых, специфических медикаментов для избавления от всех симптомов патологии не существует.
Врач назначает лечение, уменьшающее риск прогрессирования синдрома:
- препараты, корректирующие проявления почечной недостаточности, уровень белка в моче;
- мочегонные средства;
- средства, нормализующие обмен веществ в организме, восстанавливающие баланс минералов и витаминов;
- препараты для профилактики анемии.
При выраженной почечной недостаточности показано использованием процедуры гемодиализа. В тяжелых случаях развития наследственного нефрита требуется замена пораженного органа на донорский. Трансплантация почки может быть выполнена пациентам в возрасте старше 15 лет.
Детский врач
Синдром, описанный в 1927 г. A. С. Alport, характеризуется сочетанием гематурической нефропатии с врожденной глухотой. В 1954 г. Sohar (Сохар) расширяет сферу синдрома, описывая глазные аномалии, отмечающиеся в большинстве случаев. В медицинской литературе синдром появляется и под другими названиями, из которых упоминаем следующие:
- «синдром наследственной гематурии»;
- «глазо – ухо – почечный синдром»;
- «наследственный и семейный геморрагический нефрит»;
- «семейное гематурическое заболевание почек Dickinson (Дикинсон)»;
- «семейная гематурическая нефропатия, сочетающаяся с глухотой».
Этиопатогенез синдрома Альпорта. Этиология синдрома — неизвестна, но существует ряд фактов, имеющих тенденцию ее выяснить. Речь идет, вероятно, о генетической мутации (у всех больных, подвергнутых исследованию кариотипа, он был нормальным). Почечные расстройства, вероятно вызываются доминирующим геном, носимым хромосомой. Но,таким образом, представляется возможным существования «crossing over» (кроссинга). Передача почечного заболевания происходит аутосомно — доминантным образом, находящимся под влиянием пола, с большей частотой среди мальчиков, чем среди девочек. Глухота также преобладающая у мальчиков может быть вызвана рецессивной наследственностью, связанной с полом, или чрезвычайно большой проницаемостью у мужского пола.
Симптоматология синдрома Альпорта.
Нефропатия с гематурией является главной аномалией у больных, страдающих синдромом Альпорта; она более тяжелая у мужчин, чем у женщин и проявляется признаками гломерулонефрита с гематурией, цилиндрурией, альбуминурией.
Течение нефропатии хроническое и ведет к почечной недостаточности; смерть больного наступает чаще всего до 30 – летнего возраста; были описаны случаи, при которых почечная недостаточность определилась в короткое время.
Главным симптомом нефропатии при синдроме Альпорта является ранняя гематурия (появляющаяся даже у грудного ребенка), обнаруживаемая только микроскопически в результате систематического обследования больного или во время генетической анкеты (анализ мочи членов семьи, в которой был установлен диагноз синдрома Альпорта). Обычно, гематурия бывает прерывистая и появляется при ринофарингите, при мышечном напряжении и т. д.
Протеинурия может быть единственным симптомом; вначале она умеренная, но со временем становится все более выраженной. В других случаях протеинурия возникает одновременно с гематурией или следует последней. Цилиндрурия отмечается изредка. Лейкоцитурия сочетается с гематурией и протеинурией или с обеими; она всегда значительна.
Врожденная глухота поражает исключительно мужской пол. Всегда обнаруживается у больных моложе 15 лет (максимальная частота соответствует возрасту 8—10 лет); самый малый возраст, в котором обнаруживается эта аномалия — 2 года.
Речь идет о перцептивной прогрессирующей глухоте — двусторонней, как правило, асимметричной, иногда сочетающейся с поражением улиткового нерва. В большинстве случаев глухота ясно выражена клинически, проявляется значительным уменьшением слуховой перцепции; отмечаются и случаи, при которых слуховая функция кажется нормальной и только после применения аудиографического обследования устанавливается снижение слуха.
Врожденные глазные аномалии локализуются исключительно на хрусталике, а, следовательно, поражают только передний полюс глазного яблока. Речь идет о: врожденной одно- или двусторонней катаракте; о переднем или заднем, в большинстве случаев, двустороннем лентиконусе; о микросферофакии с ее немедленным последствием — близорукостью; об утолщении роговой оболочки; о нистагме.
Все эти главные аномалии появляются предпочтительно у больных мужского пола, у которых отмечается и более тяжелое течение (хотя бы только с точки зрения глазной аномалии), сравнительно с больными женского пола, пораженными той же аномалией.
Сочетающиеся (непостоянные) аномалии при синдроме Альпорта:
- запоздалое развитие веса тела и роста (явное в особенности у мальчиков) — последствие хронической нефропатии;
- слабоумие;
- пороки сердца, стеноз или недостаточность двустворчатого клапана;
- рахит со значительными изменениями конечностей и грудной клетки.
Биохимические исследования синдрома Альпорта. Биологические изменения, хотя и многочисленные, не являются специфическими и зависят от интенсивности почечной недостаточности. Помимо гематурии, альбуминурии и лейкоцитурии отмечаются и следующие изменения: повышение мочевины в крови; гипопротеинемия, вызванная тяжелой протеинурией; нормо- и гипохромная анемия; электрофорез протеинов мочи выявляет преобладающее выделение белков, ранний спад почечной концентрационной способности (понижение удельного веса мочи).
Патологическая анатомия синдрома Альпорта. Гистопатологическое исследование почки выявляет клубочковые, интерстициальные и канальцевые поражения, вначале минимальные , а затем и более выраженные у больных в возрасте старше 10 лет.
а)Клубочковые поражения. Хотя они появляются поздно (после многолетней микроскопической гематурии), по сравнению с другими почечными поражениями они являются ранними. Отмечается расширение базальной мембраны белковой баумановской капсулы и внутрикапсулярные поражения с тенденцией к гиалинизации.
б)Интерстициальные поражения имеют вид истинного интерстициального нефрита: фиброзные тяжи, лимфоцитарные инфильтраты или с пенистыми клетками, содержащими фосфолипиды и мукополисахариды.
в)Поражение канальцев обнаруживаются гораздо позже предыдущих (почти всегда у больных старше 15 лет), обусловливая атрофию или гипертрофию канальцев.
Семейный характер синдрома Альпорта был доказан большим числом семей, в которых отмечалось несколько случаев; в литературе до 1970 г. из 593 известных случаев, насчитывается 124 семейных (Cohen-Solal J. и сотр.).
Течение синдрома Альпорта. В общем, течение тяжелое у лиц мужского пола (которые редко выживают после 30-летнего возраста) и зависит от степени поражений почек и от их функциональных последствий.
У больных женского пола — течение более благоприятное и обусловлено как меньшей интенсивностью клинических проявлений нефропатии, так и непостоянством сочетания глазных и слуховых аномалий.
Прогноз Синдрома Альпорта. Сочетание нефропатии с глухотой и с глазными аномалиями, возможно, и с другими аномалиями (пороками) у мальчиков, придают течению синдрома неблагоприятный прогноз, в большинстве случаев со смертельным исходом.
Лечение синдрома Альпорта. Многочисленные врожденные пороки делают бесполезной любую терапевтическую попытку.
Похожие медицинские статьи
- Синдром Патау Синдром, описанный в 1960 г. Патау и Смиттом, известен с тех пор и под множеством других […]
- Синдром Грега (GREGG). Краснуха беременных В 1941 году Грег описал врожденные глазные аномалии в рамках коревой эмбриопатии. Позже, […]
- Синдром Ушера В 1914 году Ушер описал синдром, расположенный на грани между офтальмологией и […]
Диагностические мероприятия
В первую очередь врач изучает историю болезни родителей, так как заболевание передается от родителей к детям в 4 случаях из 5
. Он обращает внимание на следующие детали у ребенка и родителей:
- наличие гематурии;
- биопсия почек показала нарушения в структуре базальной мембраны;
- врожденные проблемы со зрением и слухом;
- в роду встречались случаи почечной недостаточности с летальным исходом;
- наблюдается постоянное снижение слуха и зрения у ребенка.
Достаточно наличия 3-х признаков
, чтобы почти наверняка поставить диагноз. Далее будут назначены дополнительные исследования в виде:
- почек,
- биопсии коллагеновых структур,
- рентгенографии,
- мочи и крови,
- консультации врача-генетика и нефролога.
Диетическое питание
Большое значение при диагнозе наследственный нефрит уделяется переходу на особый рацион питания. Диета при синдроме Альпорта предполагает полное исключение «неправильных» продуктов:
- с содержанием консервантов и ненатуральных пищевых добавок;
- острых и соленых блюд;
- жирных ингредиентов;
- с большим содержанием белка;
- любых алкогольных напитков.
В диете используются продукты, индивидуально рекомендованные врачом. Их список формируется с учетом функциональных способностей почек каждого пациента. Предлагается ввод в рацион компонентов, обеспечивающих больного необходимой энергией, витаминами и микроэлементами, а также обладающих достаточной калорийностью – телятина, нежирная говядина, птица, рыба, фрукты и овощи.
Лечение
Синдром Альпорта — неизлечимое заболевание. Замедлить развитие почечной недостаточности помогут следующие рекомендации специалистов:
- Рациональное и витаминизированное питание,
- Оптимальные физические нагрузи,
- Частые и длительные прогулки на свежем воздухе,
- Санация очагов хронической инфекции,
- Профилактика инфекционных заболеваний,
- Запрет на стандартные прививки больным детям,
- Фитосбор из крапивы, тысячелистника и черноплодной рябины показан больным детям с гематурией,
- Витаминотерапия и биостимуляторы для улучшения обмена веществ.
Правильное питание заключается в употреблении легкоусвояемых продуктов с достаточным содержанием основных нутриентов. Из рациона больных следует исключить солености и копчености, пряные и острые блюда, алкоголь, продукты с искусственными красителями, стабилизаторами, ароматизаторами. В случае нарушения функций почек необходимо ограничить потребление фосфора и кальция. Подобные рекомендации следует соблюдать больным в течение всей жизни.
Медикаментозная симптоматическая терапия:
- Для устранения гипертензии назначают ингибиторы АПФ – «Каптоприл», «Лизиноприл» и блокаторы рецепторов ангиотензина – «Лориста», «Вазотенз».
- Пиелонефрит развивается в результате присоединения инфекции. В таком случае применяют антибактериальные и противовоспалительные медикаменты.
- Для коррекции нарушений водно-электролитного обмена назначают «Фуросемид», «Верошпирон», внутривенно физраствор, глюкозу, кальция глюконат.
- Анаболические гормоны и железосодержащие препараты показаны для ускоренного образования эритроцитов.
- Иммуномодулирующая терапия – «Левамизол».
- Антигистаминные препараты – «Зиртек», «Цетрин», «Супрастин».
- Комплекс витаминов и лекарств, улучшающих обмен веществ.
Гипербарическая оксигенация оказывает положительный эффект на выраженность гематурии и функционирование почек. При переходе почечной недостаточности в терминальную стадию требуется гемодиализ и пересадка почки. Оперативное вмешательство проводится после достижения больными пятнадцатилетнего возраста. Рецидив заболевания в трансплантате не отмечается. В отдельных случаях возможно развитие нефрита.
Генотерапия синдрома в настоящее время активно разрабатывается. Ее основная цель – предупреждение и замедление ухудшения функционирования почек. Этот перспективный вариант лечения сегодня внедряется в лечебную практику западными медицинскими лабораториями.
Причины и механизм развития синдрома Альпорта
Синдром Альпорта вызван мутациями в генах COL4A4, COL4A3, COL4A5, отвечающих за биосинтез коллагена. Мутации в указанных генах нарушают нормальный синтез коллагена типа IV, который является очень важным структурным компонентом базальных мембран в почках, внутреннем ухе и глазах. Базальные мембраны – это тонкие пленочные структуры, которые поддерживают ткани и отделяют их друг от друга. При нарушении синтеза коллагена типа IV гломерулярные базальные мембраны в почках не способны нормально фильтровать токсичные продукты из крови, пропуская в мочу белки (протеинурия) и эритроциты (гематурия). Аномалии синтеза коллагена типа IV приводят к почечной недостаточности и отказу почек, что и является главной причиной смерти при синдроме Альпорта.
Синдром Альпорта
Синдром Альпорта (наследственный нефрит) — это наследственное заболевание, характеризующееся заболеваниями почек, потерей слуха и проблемами со зрением. Синдром Альпорта вызывает заболевание почек, повреждая клубочки — крошечные фильтры в почке, фильтрующие кровь.
При синдроме Альпорта поражается коллаген типа IV, который находится в клубочках, внутреннем ухе и глазах, что делает их неспособными выполнять свои функции должным образом. В свою очередь, почки становятся слабыми, а из крови отфильтровывается все меньше и меньше отходов.
Это иногда приводит к терминальной стадии почечной недостаточности.
Заболевание поражает уши, что приводит к потере слуха в раннем подростковом или позднем детстве.
У людей с синдромом Альпорта также иногда возникают проблемы со зрением, такие как неправильная форма хрусталика, что может привести к катаракте и/или близорукости.
Также иногда присутствуют белые пятнышки, разбросанные по сетчатке, называемые точечной и пятнистой ретинопатией. Однако, как правило, эти глазные пятна не приводят к слепоте.
Осложнения синдрома Альпорта встречаются чаще и тяжелее у мужчин, чем у женщин. Считается, что данный синдром диагностируется у 1 из 5000 человек.
Симптомы синдрома Альпорта
Основными симптомами синдрома Альпорта являются также его основные осложнения, такие как заболевания почек, нарушения зрения и потеря/проблемы слуха. Эти симптомы также имеют тенденцию проявляться в раннем возрасте, до официального диагноза синдрома Альпорта.
Симптомы синдрома Альпорта
- кровь в моче (гематурия). Это первый признак заболевания;
- белок в моче (протеинурия);
- артериальная гипертензия (гипертония);
- отеки в ногах, лодыжках и области глаз;
- доброкачественные гладкомышечные опухоли (лейомиоматоз);
- случайные аневризмы.
Причины синдрома Альпорта
Синдром Альпорта вызван мутациями в генах COL4A3, COL4A4 и COL4A5. Эти гены ответственны за формирование части коллагена типа IV. Коллаген является основным белком в теле, отвечающим за укрепление и поддержку соединительных тканей.
Этот коллаген типа IV действительно важен для работы почечных клубочков, и мутации в этих генах приводят к тому, что коллаген, содержащийся в клубочках, становиться аномальным. Это, в свою очередь, повреждает почки и делает их неспособными очищать кровь должным образом.
Этот коллаген содержится и в внутренних ушах, и аномалии в нем могут привести к нейросенсорной потере слуха. Коллаген типа IV также важен для поддержания формы хрусталика глаза и нормального цвета сетчатки, и именно его нарушения вызывают осложнения для глаз, связанные с синдромом.
Синдром Альпорта наследуется тремя различными способами:
Х-образный паттерн.
Это наиболее распространенный способ наследования синдрома Альпорта, и около 80 процентов людей с этим заболеванием имеют эту форму. Это вызвано мутациями в гене COL4A5.
Наследование по «Х-образному паттерну» означает, что ген расположен на Х-хромосоме.
Поскольку у мужчин есть только одна Х-хромосома, одной мутации гена на этой хромосоме достаточно, чтобы вызвать заболевание почек и другие серьезные симптомы состояние в них.
Женщины, с другой стороны, имеют две Х-хромосомы и, соответственно, две копии гена, поэтому мутация гена только в одной из хромосом обычно не может вызвать серьезные осложнения синдрома Альпорта.
Из-за этого женщины страдающие синдромом Альпорта, связанным с Х-хромосомой, обычно страдают только от крови в моче, и их иногда называют просто носителями.
Нередко у них могут развиваться другие серьезные осложнения, связанные с болезнью, но даже тогда, они их переносят проще, чем мужчины.
С Х-сцепленным типом наследования отцы не могут передать эту болезнь своим сыновьям, потому что биологически мужчины не передают свои Х-хромосомы своим детям мужского пола.
С другой стороны, каждый ребенок имеет 50-процентную вероятность наследования гена, если у матери есть дефектный ген в одной из ее Х-хромосом.
У мальчиков, наследующих дефектный ген, обычно в какой-то момент жизни развивается синдром Альпорта.
Аутосомно-доминантное наследование.
Это редкая форма наследования, и она встречается только в 5% случаев синдрома. Люди с этой формой имеют одну мутацию в генах COL4A3 или COL4A4, что означает, что только один родитель имеет аномальный ген и передаст её ребенку. При этой форме синдрома Альпорта мужчины и женщины испытывают схожие признаки с одинаковыми уровнями тяжести.
Атосомно-рецессивное наследование.
Эта форма наследования встречается примерно в 15% случаев синдрома Альпорта. Ребенок наследует болезнь таким образом только тогда, когда оба родителя являются носителями и у каждого есть копия аномального гена COL4A3 или COL4A4. При этом мужчины и женщины также страдают одинаково.
Лечение синдрома Альпорта
Не существует единого способа лечения синдрома Альпорта, потому что каждый из симптомов и осложнений лечится индивидуально.
Болезнь почек
Контролирование и замедление прогрессирования заболевания почек является первым и основным соображением при лечении синдрома Альпорта. Для этого врач может назначить:
- Ингибиторы ангиотензинпревращающего фермента (АПФ) или блокаторы рецепторов ангиотензина для снижения артериального давления и, возможно, снижения содержания белка в моче и замедления прогрессирования заболевания почек.
- Ограниченное потребление соли.
- Диуретики, также известные как мочегонные средства.
- Низкобелковую диету.
Врач, вероятно, порекомендует обратиться к диетологу, который поможет придерживаться новых ограничений для сохранения здорового питания.
Тем не менее, часто заболевание почек прогрессирует до терминальной стадии почечной недостаточности, для которой придется либо пойти на диализ, либо, в качестве альтернативы, пройти трансплантацию (пересадку) почек.
- Диализ — это искусственный процесс удаления и фильтрации отходов из организма с помощью аппарата. Аппарат для диализа в основном заменяет функцию почек.
- Пересадка почки предполагает хирургическую замену дефектной почки здоровой от донора.
Необязательно проходить диализ, прежде чем будет проведена пересадка почки, и, в конечном счете, врач поможет человеку решить, какой вариант будет лучше.
Высокое кровяное давление
Специалист назначит соответствующие таблетки/лекарства, чтобы помочь контролировать кровяное давление. Некоторые из этих препаратов — ингибиторы АПФ, бета-блокаторы и блокаторы кальциевых каналов. Они помогают снизить шансы на развитие сердечно-сосудистых заболеваний, а также замедляют прогрессирование болезни почек.
Проблемы с глазами
В случае возникновения проблем со зрением, вызванных аномалиями в форме хрусталика, врач направит пациента к офтальмологу. Далее офтальмолог может порекомендовать смену очков, выписанных по рецепту, или провести операцию по удалению катаракты. Белые пятна в глазах, возникающие в результате болезни никак не влияют на зрение, поэтому, как правило, офтальмолог на это не обращают внимания.
Потеря слуха
Если из-за синдрома Альпорта развивается потеря слуха, скорее всего, она будет постоянной. К счастью, сегодня есть большой выбор слуховых аппаратов, очень помогающих с этим.
В целом, больные также могут извлечь выгоду из изменений образа жизни, например, правильное питание, поддержание активности и здорового веса.
Заключение
Если был диагностирован синдром Альпорта, следует подробно обсудить варианты лечения с врачом, поскольку каждый отдельный случай отличается с точки зрения степени тяжести и пораженных органов. Крайне важно обратиться за помощью к специализированному врачу, который имеет опыт в лечении этого необычного заболевания.
Также следует попытаться обследовать семью с помощью генетического консультирования, чтобы определить, кто еще может подвергаться риску.
В качестве альтернативы, если вы не больны, но являетесь носителем или имеется семейная история, следует пройти генетическое консультирование, прежде чем создавать семью.
Это позволит узнать, как снизить вероятность передачи генетической мутации будущим детям, если они планируется.
Источник: https://tvojajbolit.ru/nefrologiya/sindrom_alporta/
Классификация
Выделяют три варианта наследственного нефрита
- I вариант — клинически проявляется нефритом с гематурией, тугоухостью ипоражением глаз. Течение нефрита прогрессирующее с развитием ХПН. Тип наследования — доминантный, сцепленный с Х-хромосомой. Морфологически выявляется нарушение структуры базальной мембраны, ее истончение и расщепление.
- II вариант- клинически проявляется нефритом с гематурией без тугоухости. Течение нефрита прогрессирующее с развитием хронической почечной недостаточности. Тип наследования — доминантный, сцепленный с Х-хромосомой. Морфологически выявляется истончение базальной мембраны капилляров клубочков (особенно laminadensa).
- III вариант — доброкачественная семейная гематурия. Течение благоприятное, хроническая почечная недостаточность не развивается. Тип наследования — аутосомно-доминантный или аутосомно-рецессивный. При аутосомно-рецессивном типе наследования у женщин отмечено более тяжелое течение заболевания.
[9], [10], [11], [12], [13]
Патогенез
Патогенетические звенья синдрома:
- нарушение биосинтеза коллагена или его дефицит,
- деструкция базальной мембраны почек, внутреннего уха и глазного аппарата,
- прорастание коллагеновых волокон V и VI типов,
- поражение почечных клубочков,
- иммунонегативный гломерулит,
- гиалиноз клубочков, атрофия канальцев и фиброз стромы почек,
- гломерулосклероз,
- скопление в почечной ткани липидов и липофагов,
- снижение в крови уровня Ig A, повышение IgM и G,
- снижение активности Т- и В-лимфоцитов,
- нарушение фильтрационной функции почек,
- дисфункция органа зрения и слуха,
- накопление в крови токсинов и продуктов обмена,
- протеинурия,
- гематурия,
- развитие острой почечной недостаточности,
- смерть.
Заболевание развивается постепенно с ренальных симптомов. На ранних стадиях патологии почки работают полноценно, в моче имеются следы белка, лейкоцитов и крови. Поллакиурия и никтурия сопровождаются гипертензией и другими признаками мочевого синдрома. У больных расширяются чашечки и лоханки почек, возникает аминоацидурия. Спустя некоторое время присоединяется тугоухость неврогенного происхождения.
Мужчины в наибольшей степени подвержены развитию почечной дисфункции. При отсутствии лечения смерть наступает в возрасте 15-30 лет. Женщины обычно страдают скрытой формой патологии с признаками гематурического синдрома и незначительным снижением слуха.
Классификация болезни
Выделяют 3 основных формы
заболевания:
- Доминантный тип наследования, соединенный с Х-хромосомой. Развивается истончение и расщепление базальной мембраны в почках, состоящей из коллагена. Симптомы: ухудшение слуха, снижение зрения, нефрит и гематурия. Постоянно развивается .
- Аутосомно-рецессивный тип наследования. Клиническая картина аналогична предыдущему типу, однако без ухудшения слуха.
- Аутосомно-доминантный тип наследования. Называется доброкачественной семейной гематурией. Почечная недостаточность не развивается, течение болезни благоприятное.
Симптомы болезни
Первые симптомы проявляются в возрасте 3-6 лет. Мутация генов приводит к дефициту коллагена, что в свою очередь негативно сказывается на состоянии базальной мембраны в почках, хрусталиках глаз и структуре внутреннего уха. У данных органов снижается функциональность.
В первую очередь страдают почки – ухудшается способность к фильтрации, вследствие чего в кровь начинают попадать белки, токсины и эритроциты. Развивается постоянно прогрессирующая почечная недостаточность.
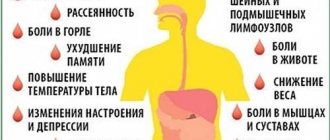
Одновременно и с задержкой происходит снижение остроты зрения и ухудшение слуха. Симптомы имеют тенденцию постоянно усиливаться и прогрессировать. У ребенка могут появляться дополнительные симптомы:
- кровь в моче;
- повышенный уровень лейкоцитов в крови и моче;
- повышенный белок в моче;
- анемия;
- симптомы интоксикации (тошнота, рвота, слабость);
- боли в мышцах;
- скачки артериального давления;
- снижение физической активности;
- головная боль;
- бессонница;
- повышение температуры тела;
- озноб;
- отставание в развитии от сверстников;
- тугоухость (неспособность отличать низкие и высокие тональности);
- аномалии хрусталика.
В дальнейшем без адекватного лечения болезнь может приобрести хроническую форму, для которой характерны:
- хроническая усталость;
- постоянное недомогание;
- сухость кожи;
- снижение аппетита;
- потеря в весе;
- неприятный вкус во рту;
- умственная отсталость и заторможенность;
- постоянная жажда и пересыхание во рту;
- бледный цвет кожи.
Диагностика
Наследственный нефрит редко диагностируется после рождения, обычно первые признаки проявляются только к 3–5 годам
Комплексное обследование детей с синдромом Альпорта включает сбор анамнеза жизни, наследственного и клинического анамнеза, изучение жалоб. Учитывая наследственную природу заболевания, значение имеют случаи почечной недостаточности у близких родственников. Для уточнения диагноза проводят ряд исследований:
- общеклинические лабораторные исследования, в том числе на синдром Альпорта;
- УЗИ почек и почечных структур, органов мочеполовой системы;
- рентгенконтрастные методы исследования (экскреторная урография);
- ДНК-тест;
- биопсия почек.
Диагностика требует дополнительной консультации нефролога, уролога, генетика, офтальмолога, кардиолога, отоларинголога. Заболевание дифференцируют от других геномных мутаций, в частности, от синдрома «кошачьего крика» — нарушения строения 5 хромосомы, когда типичные признаки выражаются в нарушении слуха и зрения еще в раннем возрасте.
Используемые источники:
- https://prosindrom.ru/genetic/sindrom-alporta.html
- https://shango.ru/sindrom-alporta-u-detei-simptomy-sindrom-alporta—bolezni-organov/
- https://lechenie-simptomy.ru/sindrom-alporta-u-detey
- https://medbe.ru/news/meditsina/sindrom-alporta-prichiny-i-simptomy-diagnostika-i-lechenie-sindroma-alporta/
- https://sindrom.info/alporta/
- https://ilive.com.ua/health/nasledstvennyy-nefrit-sindrom-alporta-u-detey_107170i15937.html
- https://m.baby.ru/wiki/sindrom-al-porta-u-detej-osobennosti-razvitia-simptomy-metody-diagnostiki-i-lecenie/
Семейный случай синдрома Альпорта у ребенка 15 лет
В последнее время наблюдается рост наследственной патологии, что связано с отсутствием контроля со стороны государства и системы здравоохранения в целом. Нет закона, запрещающего родственные браки, а отсюда и рост наследственной патологии.
Синдром Альпорта (семейный гломерулонефрит) — это редкое генетическое заболевание, которое характеризуется гломерулонефритом, прогрессирующей почечной недостаточностью, нейросенсорной тугоухостью и поражением глаз. Заболевание было впервые описано британским врачом Артуром Альпортом в 1927 году. Синдром Альпорта встречается очень редко с частотой 17 на 100 000 детского населения. Генетическая основа болезни — мутация в гене а-5 цепи коллагена IV типа. Этот тип универсален для базальных мембран почки, кохлеарного аппарата, капсулы хрусталика, сетчатки и роговицы глаза, что доказано в исследованиях с использованием моноклональных антител против этой фракции коллагена.
Согласно, литературных данных, заболевание начинается в раннем возрасте, реже дошкольном. Мы, диагностировали семейный случай синдрома Альпорта у ребенка-подростка 15 лет. Мальчик поступил на обследование в ОДММЦ города Андижана с жалобами на выраженную слабость, головную боль, пастозность лица, сильные мышечные боли в ногах, ночное недержание мочи.
Из анамнеза: ребенок от I беременности, I родов родился от молодых родителей в срок с весом 3700г. Брак неродственный. Но, в роду с обеих сторон наблюдались близкородственные браки. Беременность протекала у матери на фоне анемии средней тяжести и токсикоза II половины беременности в виде нефропатии. Прививки ребенок получил по возрасту. Перенесенные заболевания: в 8 лет перенес пиелонефрит. На учете не состоял, но при профилактических осмотрах в анализах мочи постоянно обнаруживали белок и эритроциты. Лечение по месту жительства не проводилось, так как ребенка ничего не беспокоило. С 12 лет диагностирована сенсорная тугоухость. С декабря 2020 года отметил нарушение зрения. У матери мальчика ХПН, IV стадия. Находилась в реанимационном отделении на момент госпитализации ребенка. У двоюродного брата диагностирован синдром Альпорта в 10 лет, который умер от ХПН в 13 лет. В семье ребенка в последующем диагностирован синдром Альпорта у родного брата, рожденного в 2004г, у которого заболевание проявилось в 11 лет. У сестры 2007 года рождения, заболевание проявилось в 8 лет и тоже обнаружены изменения в анализе мочи и выявлена патология со стороны органов зрения. Мать данного ребенка и ее старший брат умерли от хронической почечной недостаточности.
При осмотре: мальчик удовлетворительного питания. Физическое развитие соответствует возрасту. Вес 53кг. Состояние ребенка умеренно тяжелое. Сознание ясное. На вопросы отвечает. Положение пассивное. Выражена бледность кожных покровов и видимых слизистых. Кожа бледная, сухая. Отмечается пастозность век. Выявлены соединительнотканные стигмы: гипертелоризм, высокое нёбо, аномалии прикуса, аномальная форма ушных раковин (уши маленькие и плотно прилежат к черепу и полным отсутствием мочек), «сандалевидная щель» на стопах. Периферические лимфатические узлы не увеличены. Движения в ногах вызывают мышечную боль. Суставы спокойные. В легких везикулярное дыхание. Сердечные тоны резко приглушены, брадикардия. Пульс-60 ударов в мин, АД 110/70мм.рт.ст. Язык чистый, сосочки языка сглажены. Зев спокоен. Живот мягкий, пальпаторно безболезненный. Печень и селезенка не увеличены. Симптом Пастернацкого отрицателен с обеих сторон. Мочится мало, суточный диурез 600 мл. Стул оформленный, 1 раз в сутки.
Проведено следующее обследование: Общий анализ крови: Нв-56 г/л, эритроциты-2,5х1012, цветовой показатель- 0,5,лейк.8,7х109, палочкоядерные-2%, сегментоядерные-68%, лимфоциты -28%, моноциты-5%, эозинофилы-2%, СОЭ-30мм/ч.
В анализе мочи: соломенно-желтая, уд.вес-1015, РН-5,5, белок-0,099г/л, глюкоза-abs, эпителий почечный –ед, лейкоциты в большом количестве, эритроциты измененные — 6-8, цилиндры эритроцитарные — 2-3, соли мочевой кислоты.
Биохимия крови: мочевина — 15,7ммоль/л, остаточный азот — 58г/л, общий белок-48г/л.
УЗИ почек: правая 71х30, уменьшена в размере, контуры неровные, нечеткие, местами не дифференцированы, эхогенность повышена; левая почка 71х71см, контуры неровные, нечеткие, местами смазаны, эхогенность коркового слоя резко повышена.
Консультация окулиста: 2-х сторонняя гиперметропия, средней степени
Заключение ЛОР врача по данным аудиометрии: 2-х сторонняя тугоухость по смешанному типу II-III степени. (Рис.1)
Рисунок 1. Аудиометрия больного синдромом Альпорта.
Получил следующее лечение и рекомендации:
- Постельный режим с последующим ограничением физической нагрузки
- Диета, стол 7
- Цефтриаксон по 1,0 каждые 12 час в/м №7;
- Пиридоксин гидрохлорид по 2 мл 1 раз в сутки в/м №10;
- АТФ — по 1 мл внутримышечно через день № 10;
- Аевит по 1 капсуле в день в течение 2 недель;
- Леспенефрил по 1ч.л. 2 раза в сутки под контролем азотемии
- Эритропоэтин подкожно 2000МЕ 2 раза в неделю — 1 месяц, затем по 2000МЕ 1 раз в неделю — 1 месяц
- Альбумин 20% 100,0 внутривенно капельно №3
- Гепарин по 1500ЕД каждые 8 часов подкожно вокруг пупка под контролем свертываемости крови
- Аскорутин по 1 таб 3 раза в день за 20 минут до еды №10
В ходе проводимого лечения состояние больного в динамике временно улучшилось. Спали отеки, увеличился диурез. Исчезла головная боль, появился аппетит и настроение. Нормализовалась мочевина крови и общий анализ мочи.
Выписан под наблюдение нефролога по месту жительства и соответствующими рекомендациями. Ребенок неоднократно в последующем получал стационарное лечение в нефрологическом отделении ОДММЦ.
Таким образом, у семейных врачей, врачей педиатров должна быть настороженность в отношении наследственно обусловленных заболеваний. Диспансеризация детского населения требует более тщательного обследования с проведением комплекса необходимого исследования не только анализа крови на гемоглобин, но и общего анализа мочи, а по показаниям и более детального обследования детей. Все молодые семьи должны проходить скрининг, с целью раннего выявления наследственно обусловленной патологии.