Causes
A genetic change in the gene responsible for the process of collagen biosynthesis is the main prerequisite for the development of Alport syndrome.
A child acquires such a hereditary mutation from his parents: a daughter from her father, and from her mother, both a boy and a girl. A person may have a damaged gene, and the disease does not always develop, but will be inherited by children. One of the three genes responsible for the process of biological synthesis of type IV collagen undergoes a change. It is part of the structure of the basement membranes located in the kidneys, organs of vision and hearing. Basal membranes are special formations - thin boundaries by which some tissues of the human body are separated from each other, support and strengthen their structure. If their composition and integrity are violated, dangerous phenomena arise:
- incomplete and poor-quality filtration of toxic and other substances coming from the blood occurs;
- the composition of urine changes, in which there is a significant increase in red blood cells (hematuria or traces of blood in the urine) and unfiltered protein (proteinuria).
Such changes lead to the development of severe renal failure, in some cases to the complete cessation of organ functions and death.
Alport syndrome and its development can be triggered by several factors:
- severe illnesses caused by bacteria or viruses;
- effects on the body of vaccine components;
- excessive physical stress.
Medical statistics show that every fifth child with a confirmed diagnosis of hereditary nephritis appears in parents who do not have pathologies associated with kidney function. The cause of Alport syndrome in such cases is associated with spontaneous gene mutations.
The true causes of the disease
The true causes of alport syndrome have not yet been fully studied by scientists. In our body there is a gene whose functional responsibility is the exchange of protein in kidney tissue. So the mutation of this gene is the most likely cause of the disease.
Now let's look at the provoking factors that can contribute to the onset of the disease. These include:
- severe infectious processes;
- vaccinations;
- strong physical activity.
As can be seen from numerous cases of medical practice, sometimes the development of alport syndrome can be facilitated by a common acute respiratory viral infection. It is in view of such high risks of morbidity that children who have a family history should undergo regular diagnostic examinations more often.
An interesting fact is that Alport syndrome has a dominant inheritance. What does it mean? This suggests that if a man is a carrier of a mutated gene, then his daughters will get sick, but his sons will be born healthy. At the same time, daughters will pass the disease on to all their children.
This disease was first recorded at the beginning of the last century. A physician was observing a family that had had hematuria for several generations. Later, an association was noted between hematuria and hearing loss, as well as ocular damage. Later, when medicine improved, doctors studied more deeply the genetic nature of this syndrome.
In most cases, its “owners” have relatives with kidney pathologies and other signs of this syndrome. Consanguineous marriages also play a role, as a result of which the child increases the likelihood of receiving the same genes. In patients with Alport syndrome, changes in the immune system are detected.
Therapy methods
Treatment of Alport syndrome includes diet, medication, and timely sanitation of foci of infection.
Vaccinations for children are contraindicated; vaccination is possible only if there are strict indications.
At the moment, there are no pharmacological drugs that would affect the genetic defect.
Metabolic drugs are widely used to increase... These include cocarboxylase, vitamins A, E, B6. When protein appears in the urine, nephroprotectors (drugs that protect the kidneys) are prescribed.
- Irritable bladder syndrome in women, men and children
These include angiotensin-converting enzyme inhibitors (enalapril, lisinopril, ramipril, pyrindopril) and angiotensin receptor blockers (losartan).
The above drugs belong to the group of antihypertensive drugs.
Even with low blood pressure in children, minimal doses of medications should be taken to reduce the rate of progression of kidney failure.
Physical activity
A child with Alport syndrome should refrain from strenuous physical activity. However, he needs daily walking for at least 40 minutes.
This will improve microcirculation in the kidneys and will also promote normal development.
Dietary requirements
Should be excluded from the diet:
- salty, fatty and smoked foods;
- junk food and spicy food;
- Products with a high content of artificial colors.
It is necessary to monitor the amount of protein entering the body; if kidney failure develops, limit liquid to one liter, salt to one gram per day.
A sufficient amount of calories, vitamins, macro- and microelements must be supplied with food.
Surgical intervention
In cases where the kidneys cannot cope with the release of toxic metabolic products, a kidney transplant is performed.
- How to treat bilateral chronic pyelonephritis
Hemodialysis is performed using the kidney apparatus. The essence of the procedure is to cleanse the patient’s blood of toxic substances, which is vital.
The patient also undergoes a kidney transplant, after which immunosuppressive therapy is prescribed to prevent transplant rejection.
ethnoscience
Used in conjunction with traditional methods after consultation with your doctor. The properties of medicinal plants are used to help alleviate the clinical manifestations of the disease.
To improve microcirculation in the kidneys, you can use non-concentrated and.
Juniper fruits and birch buds will help increase the glomerular filtration rate.
Prognosis and prevention
An unfavorable prognosis is most likely for a male child, as well as in the presence of:
- high protein concentration in a general urine test;
- early development of renal disorders in close relatives;
- hearing loss.
If isolated hematuria is detected without concomitant proteinuria and hearing loss, the prognosis of the disease is favorable, functional renal failure rarely develops.
Preventive measures include pregnancy planning (remediation of chronic foci of infection, a woman should avoid excessive physical and emotional stress, register with an antenatal clinic in a timely manner, and, if indicated, undergo medical genetic counseling).
The child needs to undergo regular preventive examinations with a pediatrician. When the first signs of illness are detected, parents should inform the doctor.
Alport syndrome is a severe genetically determined disease of the kidneys, visual and hearing systems. With timely diagnosis and compliance with recommendations, it is possible to slow down the rate of development of renal failure and hearing loss.
Alport syndrome (hereditary nephritis) is rare. This pathology provokes visual impairment and hearing loss. This often leads to the need for a kidney transplant.
This syndrome appears at 3–5 years of age. Hereditary nephritis in children is constantly progressing. At the same time, the child loses hearing and vision, and the glomerular apparatus of the kidneys is affected.
Alport syndrome develops due to a mutation in the gene that produces collagen. This type of collagen is involved in the construction of the lens capsule and part of the inner ear. Hence, their function is impaired, and the functions of the kidneys themselves are also impaired.
According to the international classification, this pathology refers to congenital anomalies, chromosomal disorders and deformations. It is considered a congenital defect, since several organs and systems are affected at once.
Diagnostics
Several studies, collection and analysis of anamnesis help to make a diagnosis of hereditary nephritis. It is the genetic nature of the disease that requires studying the health characteristics of the baby’s parents and grandparents. An assessment of indicators is necessary; the presence of three of them allows one to diagnose the syndrome:
- signs of hematuria;
- kidney pathologies, renal failure, including death;
- congenital diseases of the organs of vision and hearing;
- progressive deterioration in the quality of visual and auditory perception in the child;
- the presence of changes in the structure of the basement membrane (performed during a biopsy of renal tissue).
To assess the patient’s condition and the development of the pathological process, the doctor prescribes the following studies:
- analysis for Alport syndrome (involves studying blood and urine);
- ultrasound examination of the kidneys and adrenal glands;
- X-ray examination of the organ;
- tissue biopsy;
- DNA test to determine the carrier of a mutant gene.
The patient requires additional visits to highly specialized doctors - nephrologist, geneticist, ophthalmologist, otolaryngologist.
- What is doubling of the left kidney?
Causes of hereditary diseases in children
Alport syndrome is also called hereditary kidney disease. It occurs in both boys and girls. Pathology is detected during preventive examinations.
The disease is caused by a genetic defect in protein structure. Provoking factors that lead to gene mutation include:
- Infectious disease of the mother during pregnancy. Infection is especially dangerous in the first trimester, when the formation of fetal organs and tissues occurs.
- Vaccination given to a pregnant woman.
- Excessive physical activity and emotional stress that constantly accompany the expectant mother.
Causes and development factors
The DNA molecule is a long chain of chemical compounds that underlies any gene. At the dawn of life, a person inherits many different genes. They are sorted into forty-six chromosomes. Two of them determine gender - X and Y. The cause of Alport syndrome is an incorrect gene, which contains information about the structure of type 4 collagen. There are three copies of this gene, located on the second and X chromosomes.
The Mystery of DNA - video
The incorrect structure of the gene leads to the fact that the collagen protein also has a defective configuration. First of all, this leads to changes in the vascular glomeruli. The connective tissue of the renal filter becomes inflamed over time, and hereditary nephritis is formed. With age, the glomeruli die and are replaced by scar connective tissue. Thus, the work of the kidneys in purifying the blood from harmful substances and other toxic metabolic products is significantly affected, and chronic renal failure is formed. The inflamed kidney filter begins to allow red blood cells to pass through, which give the urine a reddish tint (hematuria). Another sign of hereditary nephritis is high blood pressure. This phenomenon is caused by an excess of a special substance - angiotensin, which is released into the blood by the kidneys.
In hereditary nephritis, the kidney filter is damaged
Abnormal type 4 collagen causes the hearing organ to suffer. First of all, the transmission of sound through the labyrinth system of the inner ear is disrupted. Over time, connective tissue replaces the nerve endings responsible for producing the electrical signal. Ultimately, these changes lead to deafness.
The lens, surrounded by such collagen, often becomes cloudy with the formation of cataracts. Light through such a modified lens cannot penetrate further and reach the pigment membrane of the eye (retina). The image on it is unclear and blurry. In addition, due to a hereditary disease, the retina itself suffers, which ceases to perceive light. Because of this, black spots appear in the field of vision, which correspond to the affected areas.
The lens is the main component of the optical system of the eye
Symptoms
At three years of age and earlier, the child develops the first symptoms, expressed by isolated urinary syndrome, that is, hematuria (changes in the quantity or quality of urine, its sediment). Most often, hereditary nephritis in children is detected during an examination before entering kindergarten or during ARVI, that is, by chance. Familial nephritis differs from acquired nephritis in the absence of a time interval between the onset of the stimulus and the response.
Children do not feel discomfort at the initial stage of the disease. The main indicator of the disease is a changed color of urine; a change in pigment is always observed in the first stages. Increased pigmentation in red tones is provoked by vaccinations, fitness exercise, diseases of the larynx and lungs. At the initial stages, excess protein excretion in the urine is not constant, but proteinuria increases with the course of the disease. The presence of leukocyturia is possible - a sign of destructive changes in the body.
The next stage is the deterioration of the patient’s well-being: poisoning of the body, weakness, low blood pressure, development of deafness, and possible eye problems. A decrease in the sensitivity of the hearing aid in the initial stages is detected only through examination of the hearing aid. The most common age for the onset of deafness is between six and ten years of age. Hearing loss may be the first symptom of the disease and may even appear before urinary syndrome.
With hereditary nephritis, vision deteriorates with a twenty percent probability. Damage to vision is expressed by one of the options, such as: myopia, cataracts, changes in the physical structure of the lens.
In adolescence, a clear symptom is high blood pressure and physical underdevelopment. A characteristic stigma of the disease is a mixture of such anomalies as: malocclusion, abnormal ear shape, abnormal curvature of the little finger, a large gap between the first and second toe. Alport syndrome, acquired hereditarily, is characterized by similar abnormalities in relatives.
If we are talking about how disturbances in the structure of the kidneys occur step by step, then there are at least three stages. First: destabilization of partial functional functioning of the kidneys. Second: destructive work of both the central and middle parts of the kidneys. Third: Deterioration of glomerular filtration.
Summarizing the symptoms, we come to the conclusion that the disease passes “gradually”, that is, in stages. Initially, we are dealing with a hidden asymptomatic disease, and then it develops into an open disease with obvious symptoms.
Necessary treatment
Treatment of Alport syndrome consists of normalizing kidney function, since there are no special, specific medications to get rid of all the symptoms of the pathology.
The doctor prescribes treatment that reduces the risk of progression of the syndrome:
- drugs that correct the manifestations of renal failure, the level of protein in the urine;
- diuretics;
- means that normalize metabolism in the body, restoring the balance of minerals and vitamins;
- drugs to prevent anemia.
In case of severe renal failure, the use of hemodialysis is indicated. In severe cases of hereditary nephritis, it is necessary to replace the affected organ with a donor one. Kidney transplantation can be performed on patients over 15 years of age.
Children's doctor
The syndrome, described in 1927 by A. S. Alport, is characterized by a combination of hematuric nephropathy with congenital deafness . In 1954, Sohar expanded the scope of the syndrome, describing the ocular abnormalities observed in most cases. In the medical literature, the syndrome also appears under other names, of which we mention the following:
- “hereditary hematuria syndrome”;
- “eye – ear – renal syndrome”;
- “hereditary and familial hemorrhagic nephritis”;
- “familial hematuric kidney disease Dickinson (Dickinson)”;
- “familial hematuric nephropathy combined with deafness.”
Etiopathogenesis of Alport syndrome. The etiology of the syndrome is unknown, but there are a number of facts that tend to clarify it. This is probably a genetic mutation (in all patients who underwent karyotype examination, it was normal). Kidney disorders are probably caused by a dominant gene carried on the chromosome. But, thus, it seems possible for the existence of “crossing over”. Transmission of kidney disease occurs in an autosomal, sex-influenced dominant manner, with greater frequency among boys than among girls. Deafness, also predominant in boys, may be caused by recessive heredity associated with sex, or by extremely high permeability in the male sex.
Symptomatology of Alport syndrome.
Nephropathy with hematuria is the main abnormality in patients suffering from Alport syndrome; it is more severe in men than in women and is manifested by signs of glomerulonephritis with hematuria, cylindruria, and albuminuria.
The course of nephropathy is chronic and leads to renal failure; The death of the patient occurs most often before the age of 30; Cases have been described in which renal failure was detected in a short time.
The main symptom of nephropathy in Alport syndrome is early hematuria (appearing even in an infant), detected only microscopically as a result of a systematic examination of the patient or during a genetic questionnaire (urine analysis of family members in which the diagnosis of Alport syndrome was established). Typically, hematuria is intermittent and appears with nasopharyngitis, muscle tension, etc.
Proteinuria may be the only symptom; At first it is moderate, but over time it becomes more pronounced. In other cases, proteinuria occurs simultaneously with hematuria or follows the latter. Cylindruria is observed occasionally. Leukocyturia is combined with hematuria and proteinuria or both; it is always significant.
Congenital deafness affects only males. Always found in patients under 15 years of age (the maximum frequency corresponds to the age of 8-10 years); the youngest age at which this anomaly is detected is 2 years.
We are talking about progressive perceptual deafness - bilateral, usually asymmetric, sometimes combined with damage to the cochlear nerve. In most cases, deafness is clearly expressed clinically, manifested by a significant decrease in auditory perception; There are also cases in which the auditory function seems normal and only after the use of an audiographic examination is hearing loss established.
Congenital ocular anomalies are localized exclusively on the lens and, therefore, affect only the anterior pole of the eyeball. We are talking about: congenital unilateral or bilateral cataracts; about anterior or posterior, in most cases, bilateral lenticonus; about microspherophakia with its immediate consequence - myopia; about thickening of the cornea; about nystagmus.
All these main anomalies appear preferentially in male patients , who also have a more severe course (if only from the point of view of the ocular anomaly), compared with female patients affected by the same anomaly.
Associated (non-constant) anomalies in Alport syndrome:
- delayed development of body weight and height (obvious especially in boys) is a consequence of chronic nephropathy;
- dementia;
- heart defects, stenosis or insufficiency of the bicuspid valve;
- rickets with significant changes in the limbs and chest.
Biochemical studies of Alport syndrome. Biological changes, although numerous, are not specific and depend on the intensity of renal failure. In addition to hematuria, albuminuria and leukocyturia, the following changes are also noted: increased urea in the blood; hypoproteinemia caused by severe proteinuria; normo- and hypochromic anemia; electrophoresis of urine proteins reveals the predominant release of proteins, an early decline in renal concentrating ability (decrease in the specific gravity of urine).
Pathological anatomy of Alport syndrome. Histopathological examination of the kidney reveals glomerular, interstitial and tubular lesions, initially minimal and then more pronounced in patients over 10 years of age.
a) Glomerular lesions. Although they appear late (after many years of microscopic hematuria), compared with other renal lesions they are early. There is an expansion of the basement membrane of the protein Bauman capsule and intracapsular lesions with a tendency to hyalinization.
b) Interstitial lesions have the appearance of true interstitial nephritis: fibrous strands, lymphocytic infiltrates or with foam cells containing phospholipids and mucopolysaccharides.
c) Lesions of the tubules are detected much later than the previous ones (almost always in patients over 15 years old), causing atrophy or hypertrophy of the tubules.
The familial nature of Alport syndrome has been proven by a large number of families in which several cases were noted; in the literature before 1970, out of 593 known cases, there were 124 family ones (Cohen-Solal J. et al.).
The course of Alport syndrome. In general, the course is severe in males (who rarely survive beyond 30 years of age) and depends on the extent of kidney damage and its functional consequences.
In female patients, the course is more favorable and is due to both the lower intensity of the clinical manifestations of nephropathy and the variability of the combination of ocular and auditory anomalies.
Prognosis of Alport Syndrome. The combination of nephropathy with deafness and eye anomalies, possibly with other anomalies (defects) in boys, gives the course of the syndrome an unfavorable prognosis, in most cases with a fatal outcome.
Treatment of Alport syndrome. Numerous congenital defects render any therapeutic attempt futile.
Related medical articles
- Patau Syndrome The syndrome, described in 1960 by Patau and Smith, has since been known under many other […]
- Greg's syndrome (GREGG). Rubella of pregnancy In 1941, Greg described congenital ocular abnormalities under the umbrella of measles embryopathy. Later, […]
- Usher's Syndrome In 1914, Usher described a syndrome that straddles the line between ophthalmology and […]
Diagnostic measures
First of all, the doctor studies the medical history of the parents, since the disease is transmitted from parents to children in 4 cases out of 5
. He pays attention to the following details in the child and parents:
- presence of hematuria;
- a kidney biopsy showed abnormalities in the structure of the basement membrane;
- congenital problems with vision and hearing;
- there were cases of renal failure with a fatal outcome in the family;
- There is a constant decrease in hearing and vision in the child.
It is enough to have 3 signs
to almost certainly make a diagnosis. Further additional studies will be prescribed in the form of:
- kidney,
- biopsy of collagen structures,
- radiography,
- urine and blood,
- consultations with a geneticist and nephrologist.
Diet food
When diagnosing hereditary nephritis, great importance is given to switching to a special diet. The Alport syndrome diet involves the complete exclusion of “wrong” foods:
- containing preservatives and unnatural food additives;
- spicy and salty dishes;
- fatty ingredients;
- high in protein;
- any alcoholic drinks.
The diet uses products individually recommended by a doctor. Their list is formed taking into account the functional abilities of the kidneys of each patient. It is proposed to introduce into the diet components that provide the patient with the necessary energy, vitamins and microelements, as well as those with sufficient calorie content - veal, lean beef, poultry, fish, fruits and vegetables.
Treatment
Alport syndrome is an incurable disease. The following expert recommendations will help slow down the development of kidney failure:
- Rational and fortified diet,
- Optimal physical activity
- Frequent and long walks in the fresh air,
- Sanitation of foci of chronic infection,
- Prevention of infectious diseases,
- Ban on routine vaccinations for sick children
- A herbal collection of nettle, yarrow and chokeberry is indicated for sick children with hematuria,
- Vitamin therapy and biostimulants to improve metabolism.
Proper nutrition consists of eating easily digestible foods with sufficient amounts of essential nutrients. From the diet of patients, salted and smoked foods, spicy and hot dishes, alcohol, and products with artificial colors, stabilizers, and flavors should be excluded. In case of impaired renal function, it is necessary to limit the intake of phosphorus and calcium. Such recommendations should be followed by patients throughout their lives.
Drug symptomatic therapy:
- To eliminate hypertension, ACE inhibitors are prescribed - Captopril, Lisinopril and angiotensin receptor blockers - Lorista, Vasotens.
- Pyelonephritis develops as a result of infection. In this case, antibacterial and anti-inflammatory medications are used.
- To correct disturbances in water-electrolyte metabolism, Furosemide, Veroshpiron, intravenous saline, glucose, and calcium gluconate are prescribed.
- Anabolic hormones and iron-containing drugs are indicated to accelerate the formation of red blood cells.
- Immunomodulatory therapy - Levamisole.
- Antihistamines - Zirtec, Cetrin, Suprastin.
- A complex of vitamins and medications that improve metabolism.
Hyperbaric oxygen therapy has a positive effect on the severity of hematuria and kidney function. When renal failure progresses to the terminal stage, hemodialysis and a kidney transplant are required. Surgery is performed after the patient reaches the age of fifteen. There is no recurrence of the disease in the graft. In some cases, nephritis may develop.
Gene therapy for the syndrome is currently being actively developed. Its main goal is to prevent and slow down the deterioration of kidney function. This promising treatment option is now being introduced into medical practice by Western medical laboratories.
Causes and mechanism of development of Alport syndrome
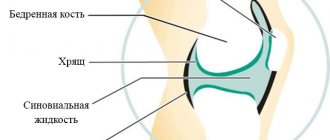
Alport syndrome is caused by mutations in the COL4A4, COL4A3, COL4A5 genes, which are responsible for collagen biosynthesis. Mutations in these genes disrupt the normal synthesis of type IV collagen, which is a very important structural component of the basement membranes in the kidneys, inner ear and eyes. Basement membranes are thin film-like structures that support tissues and separate them from each other. When type IV collagen synthesis is impaired, the glomerular basement membranes in the kidneys are unable to properly filter toxic products from the blood, allowing proteins (proteinuria) and red blood cells (hematuria) to pass into the urine. Abnormalities in the synthesis of type IV collagen lead to renal failure and kidney failure, which is the main cause of death in Alport syndrome.
Alport syndrome
Alport syndrome (hereditary nephritis) is an inherited disease characterized by kidney disease, hearing loss, and vision problems. Alport syndrome causes kidney disease by damaging the glomeruli, the tiny filters in the kidney that filter the blood.
Alport syndrome affects type IV collagen, which is found in the glomeruli, inner ear and eyes, making them unable to perform their functions properly. In turn, the kidneys become weak and less and less waste is filtered from the blood.
This sometimes leads to end-stage renal failure.
The disease affects the ears, leading to hearing loss in early adolescence or late childhood.
People with Alport syndrome also sometimes have vision problems, such as an abnormally shaped lens, which can lead to cataracts and/or nearsightedness.
There are also sometimes white spots scattered across the retina, called punctate and macular retinopathy. However, these eyespots generally do not cause blindness.
Complications of Alport syndrome are more common and severe in men than in women. It is believed that this syndrome is diagnosed in 1 in 5,000 people.
Symptoms of Alport syndrome
The main symptoms of Alport syndrome are also its main complications, such as kidney disease, visual impairment and hearing loss/problems. These symptoms also tend to appear early in life, before Alport syndrome is formally diagnosed.
Symptoms of Alport syndrome
- blood in the urine (hematuria). This is the first sign of the disease;
- protein in the urine (proteinuria);
- arterial hypertension (hypertension);
- swelling in the legs, ankles, and eye area;
- benign smooth muscle tumors (leiomyomatosis);
- occasional aneurysms.
Causes of Alport syndrome
Alport syndrome is caused by mutations in the COL4A3, COL4A4 and COL4A5 genes. These genes are responsible for the formation of part of type IV collagen. Collagen is the main protein in the body responsible for strengthening and supporting connective tissues.
This type IV collagen is really important for the function of the glomeruli, and mutations in these genes cause the collagen contained in the glomeruli to become abnormal. This in turn damages the kidneys and makes them unable to cleanse the blood properly.
This collagen is also found in the inner ears, and abnormalities in it can lead to sensorineural hearing loss. Type IV collagen is also important for maintaining the shape of the eye's lens and the normal color of the retina, and it is its disruption that causes the eye complications associated with the syndrome.
Alport syndrome is inherited in three different ways:
X-shaped pattern.
This is the most common way Alport syndrome is inherited, and about 80 percent of people with the condition have this form. It is caused by mutations in the COL4A5 gene.
“X-pattern” inheritance means that the gene is located on the X chromosome.
Since males have only one X chromosome, one gene mutation on that chromosome is enough to cause kidney disease and other serious symptoms in them.
Women, on the other hand, have two X chromosomes and therefore two copies of the gene, so a gene mutation in only one of the chromosomes usually cannot cause serious complications of Alport syndrome.
Because of this, women with X-linked Alport syndrome typically suffer only from blood in the urine and are sometimes simply called carriers.
Often they may develop other serious complications associated with the disease, but even then, they tolerate them more easily than men.
With X-linked inheritance, fathers cannot pass the disease on to their sons because biologically males do not pass on their X chromosomes to their male children.
On the other hand, each child has a 50 percent chance of inheriting the gene if the mother has the defective gene on one of her X chromosomes.
Boys who inherit the defective gene usually develop Alport syndrome at some point in their lives.
Autosomal dominant inheritance.
This is a rare form of inheritance and occurs in only 5% of cases of the syndrome. People with this form have one mutation in the COL4A3 or COL4A4 genes, which means only one parent has the abnormal gene and will pass it on to the child. In this form of Alport syndrome, men and women experience similar symptoms with similar levels of severity.
Atosomal recessive inheritance.
This form of inheritance occurs in approximately 15% of cases of Alport syndrome. A child inherits the disease in this way only when both parents are carriers and each has a copy of the abnormal COL4A3 or COL4A4 gene. At the same time, men and women also suffer equally.
Treatment of Alport syndrome
There is no single treatment for Alport syndrome because each of the symptoms and complications is treated individually.
Kidney disease
Controlling and slowing the progression of kidney disease is the first and foremost consideration in the treatment of Alport syndrome. To do this, the doctor may prescribe:
- Angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers to lower blood pressure and possibly reduce protein in the urine and slow the progression of kidney disease.
- Limited salt intake.
- Diuretics, also known as diuretics.
- Low protein diet.
Your doctor will likely recommend seeing a dietitian to help you adhere to your new restrictions to maintain a healthy diet.
However, often kidney disease progresses to end-stage renal failure, which will require either going on dialysis or, alternatively, undergoing a kidney transplant (transplant).
- Dialysis is an artificial process of removing and filtering waste from the body using a machine. The dialysis machine basically replaces the function of the kidneys.
- A kidney transplant involves surgically replacing a defective kidney with a healthy one from a donor.
It is not necessary to undergo dialysis before a kidney transplant, and ultimately the doctor will help the person decide which option is best.
High blood pressure
The specialist will prescribe appropriate tablets/medicines to help control your blood pressure. Some of these drugs are ACE inhibitors, beta blockers, and calcium channel blockers. They help reduce your chances of developing cardiovascular disease and also slow the progression of kidney disease.
Eye problems
In case of vision problems caused by abnormalities in the shape of the lens, the doctor will refer the patient to an ophthalmologist. Next, your ophthalmologist may recommend changing your prescription glasses or performing cataract surgery. White spots in the eyes resulting from the disease do not affect vision in any way, so, as a rule, the ophthalmologist does not pay attention to this.
Hearing loss
If hearing loss develops due to Alport syndrome, it is likely to be permanent. Fortunately, today there are a large selection of hearing aids that can help greatly with this.
In general, sufferers can also benefit from lifestyle changes, such as eating right, staying active and maintaining a healthy weight.
Conclusion
If Alport syndrome has been diagnosed, treatment options should be discussed in detail with your doctor, as each individual case differs in terms of severity and organs affected. It is extremely important to seek help from a specialized doctor who has experience in treating this unusual condition.
You should also try to screen the family through genetic counseling to determine who else may be at risk.
Alternatively, if you are not affected but are a carrier or have a family history, you should undergo genetic counseling before starting a family.
This will allow you to find out how to reduce the likelihood of passing on a genetic mutation to future children, if they are planned.
Source: https://tvojajbolit.ru/nefrologiya/sindrom_alporta/
Classification
There are three types of hereditary nephritis
- Option I - clinically manifested by nephritis with hematuria, hearing loss and eye damage. The course of nephritis is progressive with the development of chronic renal failure. The type of inheritance is dominant, linked to the X chromosome. Morphologically, a violation of the structure of the basement membrane, its thinning and splitting is revealed.
- Option II - clinically manifested by nephritis with hematuria without hearing loss. The course of nephritis is progressive with the development of chronic renal failure. The type of inheritance is dominant, linked to the X chromosome. Morphologically, thinning of the basement membrane of the glomerular capillaries (especially laminadensa) is revealed.
- Option III - benign familial hematuria. The course is favorable, chronic renal failure does not develop. The type of inheritance is autosomal dominant or autosomal recessive. With an autosomal recessive type of inheritance, women have a more severe course of the disease.
[9], [10], [11], [12], [13]
Pathogenesis
Pathogenetic links of the syndrome:
- violation of collagen biosynthesis or its deficiency,
- destruction of the basement membrane of the kidneys, inner ear and ocular apparatus,
- sprouting of collagen fibers types V and VI,
- damage to the renal glomeruli,
- immunonegative glomerulitis,
- glomerular hyalinosis, tubular atrophy and renal stromal fibrosis,
- glomerulosclerosis,
- accumulation of lipids and lipophages in the renal tissue,
- decrease in the blood level of Ig A, increase in IgM and G,
- decreased activity of T- and B-lymphocytes,
- impaired renal filtration function,
- dysfunction of the organ of vision and hearing,
- accumulation of toxins and metabolic products in the blood,
- proteinuria,
- hematuria,
- development of acute renal failure,
- death.
The disease develops gradually with renal symptoms. In the early stages of pathology, the kidneys work fully, and there are traces of protein, leukocytes and blood in the urine. Pollakiuria and nocturia are accompanied by hypertension and other signs of urinary syndrome. In patients, the calyces and pelvis of the kidneys dilate, and aminoaciduria occurs. After some time, hearing loss of neurogenic origin occurs.
Men are most susceptible to developing kidney dysfunction. If left untreated, death occurs between 15 and 30 years of age. Women usually suffer from a latent form of pathology with signs of hematuria syndrome and slight hearing loss.
Classification of the disease
There are 3 main forms
diseases:
- Dominant type of inheritance associated with the X chromosome. Thinning and splitting of the basement membrane in the kidneys, consisting of collagen, develops. Symptoms: hearing loss, decreased vision, nephritis and hematuria. Constantly evolving.
- Autosomal recessive type of inheritance. The clinical picture is similar to the previous type, but without hearing impairment.
- Autosomal dominant type of inheritance. Called benign familial hematuria. Renal failure does not develop, the course of the disease is favorable.
Symptoms of the disease
The first symptoms appear at the age of 3-6 years . Gene mutation leads to collagen deficiency, which in turn negatively affects the condition of the basement membrane in the kidneys, eye lenses and the structure of the inner ear. The functionality of these organs decreases.
The kidneys are the first to suffer - their filtration ability deteriorates, as a result of which proteins, toxins and red blood cells begin to enter the blood. Constantly progressive renal failure develops.
At the same time and with a delay, a decrease in visual acuity and deterioration of hearing occur. Symptoms tend to get worse and worse . Your child may experience additional symptoms:
- blood in urine;
- increased levels of leukocytes in the blood and urine;
- increased protein in the urine;
- anemia;
- symptoms of intoxication (nausea, vomiting, weakness);
- muscle pain;
- blood pressure surges;
- decreased physical activity;
- headache;
- insomnia;
- increased body temperature;
- chills;
- developmental delay from peers;
- hearing loss (inability to distinguish between low and high tones);
- lens abnormalities.
In the future, without adequate treatment, the disease may acquire a chronic form , which is characterized by:
- chronic fatigue;
- constant malaise;
- dry skin;
- decreased appetite;
- weight loss;
- unpleasant taste in the mouth;
- mental retardation and lethargy;
- constant thirst and dry mouth;
- pale skin color.
Diagnostics
Hereditary nephritis is rarely diagnosed after birth, usually the first signs appear only by 3–5 years of age.
A comprehensive examination of children with Alport syndrome includes collecting a life history, hereditary and clinical history, and studying complaints. Given the hereditary nature of the disease, cases of renal failure in close relatives are important. To clarify the diagnosis, a number of studies are carried out:
- general clinical laboratory tests, including Alport syndrome;
- Ultrasound of the kidneys and renal structures, organs of the genitourinary system;
- X-ray contrast research methods (excretory urography);
- DNA test;
- kidney biopsy.
Diagnosis requires additional consultation with a nephrologist, urologist, geneticist, ophthalmologist, cardiologist, and otolaryngologist. The disease is differentiated from other genomic mutations, in particular, from the “cry of the cat” syndrome - a disorder of the structure of chromosome 5, when typical signs are expressed in hearing and vision impairment at an early age.
Sources used:
- https://prosindrom.ru/genetic/sindrom-alporta.html
- https://shango.ru/sindrom-alporta-u-detei-simptomy-sindrom-alporta—bolezni-organov/
- https://lechenie-simptomy.ru/sindrom-alporta-u-detey
- https://medbe.ru/news/meditsina/sindrom-alporta-prichiny-i-simptomy-diagnostika-i-lechenie-sindroma-alporta/
- https://sindrom.info/alporta/
- https://ilive.com.ua/health/nasledstvennyy-nefrit-sindrom-alporta-u-detey_107170i15937.html
- https://m.baby.ru/wiki/sindrom-al-porta-u-detej-osobennosti-razvitia-simptomy-metody-diagnostiki-i-lecenie/
A familial case of Alport syndrome in a 15-year-old child
Recently, there has been an increase in hereditary pathology, which is due to the lack of control by the state and the health care system as a whole. There is no law prohibiting consanguineous marriages, and hence the increase in hereditary pathology.
Alport syndrome (familial glomerulonephritis) is a rare genetic disorder characterized by glomerulonephritis, progressive renal failure, sensorineural hearing loss, and eye damage. The disease was first described by British physician Arthur Alport in 1927. Alport syndrome is very rare with an incidence of 17 per 100,000 children. The genetic basis of the disease is a mutation in the a-5 gene of the type IV collagen chain. This type is universal for the basement membranes of the kidney, cochlear apparatus, lens capsule, retina and cornea, as proven in studies using monoclonal antibodies against this collagen fraction.
According to literature data, the disease begins at an early age, less often preschool. We diagnosed a familial case of Alport syndrome in a 15-year-old teenage child. The boy was admitted for examination to the Children's Medical Center in Andijan with complaints of severe weakness, headache, facial pastiness, severe muscle pain in the legs, and bedwetting.
From the anamnesis: a child from the first pregnancy, first birth was born to young parents at term with a weight of 3700 g. Unrelated marriage. But there were consanguineous marriages on both sides of the family. The mother's pregnancy proceeded against the background of moderate anemia and toxicosis of the second half of pregnancy in the form of nephropathy. The child received vaccinations according to age. Previous illnesses: at the age of 8 he suffered from pyelonephritis. He was not registered, but during routine examinations, protein and red blood cells were constantly detected in urine tests. Treatment at the place of residence was not carried out, since nothing bothered the child. Sensory hearing loss was diagnosed at the age of 12. Since December 2020, he has noticed visual impairment. The boy's mother has stage IV chronic renal failure. She was in the intensive care unit at the time the child was hospitalized. A cousin was diagnosed with Alport syndrome at age 10 and died of chronic renal failure at age 13. In the child's family, Alport syndrome was subsequently diagnosed in a sibling born in 2004, in whom the disease manifested itself at the age of 11. In my sister, born in 2007, the disease manifested itself at the age of 8 and changes were also detected in urine analysis and pathology in the organs of vision was detected. The mother of this child and her older brother died of chronic renal failure.
On examination: the boy has satisfactory nutrition. Physical development corresponds to age. Weight 53kg. The child's condition is moderately serious. Consciousness is clear. Answers questions. The position is passive. Pronounced pallor of the skin and visible mucous membranes. The skin is pale, dry. Pasty eyelids are noted. Connective tissue stigmata were identified: hypertelorism, high palate, malocclusion, abnormal shape of the ears (ears are small and tightly attached to the skull and a complete absence of lobes), “sandal-shaped gap” on the feet. Peripheral lymph nodes are not enlarged. Movement in the legs causes muscle pain. The joints are calm. Vesicular breathing in the lungs. Heart sounds are sharply muffled, bradycardia. Pulse 60 beats per minute, blood pressure 110/70 mm Hg. The tongue is clean, the tongue papillae are smoothed. Zev is calm. The abdomen is soft and painless on palpation. The liver and spleen are not enlarged. Pasternatsky's symptom is negative on both sides. He urinates little, daily diuresis is 600 ml. The chair is decorated, 1 time per day.
The following examination was carried out: General blood test: Hb-56 g/l, erythrocytes - 2.5x1012, color index - 0.5, leukemia - 8.7x109, band cells - 2%, segmented cells - 68%, lymphocytes - 28%, monocytes -5%, eosinophils -2%, ESR -30mm/h.
In urine analysis: straw-yellow, specific weight - 1015, pH - 5.5, protein - 0.099 g/l, glucose-abs, renal epithelium - units, leukocytes in large numbers, altered red blood cells - 6-8, erythrocyte casts - 2-3, uric acid salts.
Blood biochemistry: urea - 15.7 mmol/l, residual nitrogen - 58 g/l, total protein - 48 g/l.
Ultrasound of the kidneys: right 71x30, reduced in size, uneven, unclear contours, not differentiated in places, increased echogenicity; the left kidney is 71x71cm, the contours are uneven, unclear, blurred in places, the echogenicity of the cortical layer is sharply increased.
Consultation with an ophthalmologist: 2-sided hypermetropia, moderate
Conclusion of an ENT doctor based on audiometry data: 2-sided hearing loss of mixed type II-III degree. (Fig.1)
Figure 1. Audiometry of a patient with Alport syndrome.
Received the following treatment and recommendations:
- Bed rest followed by limitation of physical activity
- Diet, table 7
- Ceftriaxone 1.0 every 12 hours IM No. 7;
- Pyridoxine hydrochloride 2 ml 1 time per day IM No. 10;
- ATP - 1 ml intramuscularly every other day No. 10;
- Aevit 1 capsule per day for 2 weeks;
- Lespenefril 1 tsp. 2 times a day under the control of azotemia
- Erythropoietin subcutaneously 2000IU 2 times a week - 1 month, then 2000IU 1 time per week - 1 month
- Albumin 20% 100.0 intravenous drip No. 3
- Heparin 1500 units every 8 hours subcutaneously around the navel under control of blood clotting
- Ascorutin 1 tablet 3 times a day 20 minutes before meals No. 10
During the course of treatment, the patient's condition temporarily improved. The swelling subsided and diuresis increased. The headache disappeared, appetite and mood appeared. Blood urea and general urine analysis returned to normal.
He was discharged under the supervision of a nephrologist at his place of residence and with appropriate recommendations. The child repeatedly subsequently received inpatient treatment in the nephrology department of the Children's Medical Center.
Thus, family doctors and pediatricians should be wary of hereditary diseases. Clinical examination of the children's population requires a more thorough examination with a complex of necessary studies, not only a blood test for hemoglobin, but also a general urine test, and, if indicated, a more detailed examination of children. All young families should undergo screening in order to early detect hereditary pathology.