Etiopathogenesis
Neurofibromatosis type I was the first tumor disease with a proven genetic origin.
The locus of genes, the breakdown of which leads to the development of neurofibromatosis, is located on the long arm of chromosome 17 (17q11.2). It consists of 400 thousand nucleotide pairs. It contains information responsible for the synthesis of one of the components of myelin glycoprotein, neurofibromin and other proteins. In neurofibromatosis type I, various types of mutations and rearrangements are noted at this locus - translocations, deletions, inversions and point mutations. The nature of the mutations is very specific: more than 80% of them lead to the synthesis of a non-functional “truncated” protein or to the complete absence of a transcript (nonsense mutations, mutations at splice sites, frameshift deletions and insertions, large deletions covering the entire gene or a significant part of it ). Neurofibromin is a cytoplasmic protein consisting of 2818 amino acids. It is involved in the inactivation of promoter proteins (RAS protein and its analogues), providing dynamic control of cell growth. The NF-1 gene is one of the main tumor suppressor genes for approximately half of the body's tissues, primarily of neuroectodermal origin, the proliferation of which is determined by the RAS protein system. Neurofibromin also affects the content of adenosine monophosphate (AMP) in the cell. AMP, in turn, indirectly inhibits the processes of cell division.
When the NF1 gene is damaged in one of the chromosomes of the 17th pair, half of the synthesized neurofibromin becomes defective, and a shift in the equilibrium of cell growth towards proliferation is noted. The remaining intact allelic (located on a paired chromosome) NF1 gene ensures the synthesis of normal neurofibromin. The severity of clinical manifestations of neurofibromatosis is determined by the state of general antitumor immunity and can vary widely. Benign neoplasms appear.
In case of loss due to mutation of the allelic normal NF1 gene, rapid uncontrolled cell growth occurs, that is, a malignant tumor develops (most often neurofibrosarcoma or neuroblastoma). The probability of their occurrence is 3-15%. The probability of developing a malignant tumor associated with neurofibromatosis type I is hundreds of times higher than that in the population (200-500 times only for myeloid leukemia).
Diagnosis and testing for neurofibromatosis
Neurofibromatosis
Neurofibromatosis is detected through a physical examination of the tumor and a complete review of the patient's medical history.
During the examination, increased attention should be paid to looking for changes in the appearance of the skin and the presence of tumors or bone pathology. It may be necessary to screen close blood relatives (including parents, siblings and children) for signs of neurofibromatosis.
Diagnostic Imaging
The most suitable means for treatment are instruments such as, which are used to examine body tissues and search for tumors when they are still barely noticeable. If tumors on the nerves of the eyes and ears are detected early, the chance of successful treatment increases.
Symptoms of Recklinghausen's disease
Recognize and treat psoriatic arthritis, its symptoms and treatments
The first symptoms of the hereditary disease in question are detected in a newborn child - multiple pigment spots of small size and oval shape are detected on the skin. In most cases they are yellowish-brown in color, but in some cases blue, purple, or depigmented patches may be present.
Hyperpigmentation most often forms in the armpits, inguinal folds and on the torso. Much less often, such spots are found on the neck, upper or lower extremities, and face. It is characteristic that with age the number of such pronounced pigment spots increases, and their size also grows.
The progression of the disease leads to the appearance of subcutaneous and cutaneous neurofibromas, which are hernial-type protrusions up to several centimeters in size. Most often, such formations appear at the age of 20 years and older, the color of the skin over them either remains unchanged or becomes brown, pinkish-blue. Most often, such tumors form on the surface of the body; upon palpation, their soft consistency is determined (in rare cases, it can be dense), and when you try to press on the tumor with your finger, it “falls” into the void.
In some cases, there is hair growth at the top of the neurofibroma, in which case the tumor will be called a plexiform fibroma. Such formations have a massive sac-like appearance; winding nerve trunks can be felt in them. Plexiform fibromas can be located on the surface of the skin or paraspinal, in the mediastinum and in the retroperitoneum. If such fibromas are located deep in the body, then it will be easy to determine their location - in the projection on the skin, pigment spots will be visible, on the surface of which hair grows.
Important! It is plexiform fibromas that more often undergo malignant transformation and form into neurofibrosarcomas. The formation of neurofibromas may be accompanied by sensory disturbances, numbness, itching, tingling, and painful sensations
All pathological changes described above will be observed in almost all organs, but most often in the endocrine, bone, nervous systems and visual organs
The formation of neurofibromas can be accompanied by sensory disturbances, numbness, itching, tingling, and painful sensations. All the pathological changes described above will be observed in almost all organs, but most often in the endocrine, bone, nervous systems and organs of vision.
Doctors note that there are cases when Recklinghausen's disease is accompanied by the formation of neuromas, melanotic spots on the palms and soles of the feet, pseudoatrophic and bluish-blue spots.
The peripheral form of the hereditary disease in question is accompanied by the development of multiple neurofibromas and neurinomas, and if they are located on large branches of the nerve trunks, then a violation of lymphatic drainage may occur, which is manifested by elephantiasis. Depending on where the tumor is located, the patient’s legs, tongue, arms, and parts of the face may become enlarged. In addition, the peripheral form of Ricklenhausen's disease in almost every case is accompanied by anomalies of the skeletal system - for example, spina bifida, asymmetry of the skull and scoliosis. Specific signs of the disease in question include the appearance of pigmented hamartros in the iris (whitish spots) - diagnosed in 94% of all patients.
The central form of Recklinghausen disease is characterized by the formation of gliomas and meningiomas on the spinal roots or cranial nerves. Symptoms of this form of the disease in question depend on the location of the tumors, but in most cases patients present the following complaints:
- balance disorders;
- headache;
- speech changes;
- facial distortion and other symptoms of damage to the craniofacial nerves;
- loss of sensitivity.
It is with the central form of Recklinghausen's disease that patients are often diagnosed with acoustic neuromas, which lead to bilateral deafness; in some patients, lesions of the membranes of the eye are detected, which leads to the development of glaucoma and tumors of the optic nerve.
In addition to the above symptoms, Recklinghausen's neurofibromatosis is accompanied by concomitant pathologies - for example, mental retardation, early puberty, epileptic seizures, gynecomastia.
Clinical picture
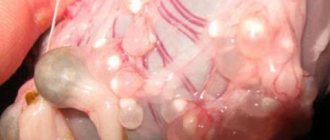
Macroscopic specimen of a neurofibroma Histological specimen of a plexiform neurofibroma
Neurofibromatosis type I is manifested by a number of pathognomonic symptoms. These include the presence of pigment spots on the café-au-lait skin, neurofibromas, most of which are located superficially on the skin, Lisch nodules - hamartomas of the iris[25].
Neurofibromatosis type I often begins with scoliosis (curvature of the spine), followed by learning difficulties, vision problems and epilepsy.
Neurofibromas are most often localized along the peripheral nerves. However, the spinal cord and brain can be affected; neurofibromas are found on the eyelids, conjunctiva, mediastinum, and abdominal cavity. Depending on the location, neurofibromas can cause various clinical symptoms: seizures, dysfunction of cranial nerves and segments of the spinal cord, paralysis of the eye muscles, ptosis, compression of the mediastinal organs.
Neurofibromas
Main article: Neurofibroma
Multiple cutaneous neurofibromas on the back of a patient with neurofibromatosis type I
This disease is characterized by the appearance of a large number of neurofibromas, both cutaneous and plexiform. Cutaneous neurofibromas are small, benign and limited neoplasms. They are located subcutaneously, growing on the membranes of small nerves of the skin. Plexiform neurofibromas develop on large nerves and lead to impairment of their functions[26]. Also, plexiform neurofibromas are characterized by their large size. Occurs in 30% of patients with neurofibromatosis type I[22].
Clinically, nerve damage manifests as chronic pain, numbness and/or muscle paralysis.
Tumors of the central nervous system
Optic nerve glioma
In neurofibromatosis type I, the incidence of tumors of the central nervous system ranges from 5[27] to 30%[28][29]. In many cases, CNS tumors in patients with neurofibromatosis are not detected[30]. For the first time[30], the relationship between neurofibromatosis type I and intracranial neoplasms was noted in 1940[31].
The most common CNS tumors associated with this disease are optic nerve gliomas, astrocytomas, ependymomas, acoustic neuromas, meningiomas and neurofibromas[32][33][34][35].
The clinical picture for tumors of the central nervous system will depend on their size, location and formations involved in the pathological process.
Pigment disorders
For neurofibromatosis, pigment spots from light beige to dark brown are pathognomonic, which are detected on the skin of the trunk and limbs, less often on the face, neck, and oral mucosa. They have a smooth surface and do not protrude above the skin level. Histological examination of pigment spots reveals a diffuse accumulation in the papillary layer of the dermis of melanoblasts and melanocytes with inclusions of melanin in the cytoplasm [36].
These pigment spots are in the nature of “coffee with milk” spots (French café-au-lait, English milk coffee) and “freckled clusters” [24]. In some cases, the spots are blue or purple in color, and depigmentation is less common[37].
Lisch nodules
Lisch nodules (iris hamartoma)
Lisch nodules occur in almost all patients with neurofibromatosis type I over 20 years of age. They appear as small whitish spots (hamartomas) on the iris of the eye. Lisch nodules are not visible to the naked eye; an ophthalmological examination is necessary. The detection of Lisch nodules increases with the age of the patient: at the age of 0 to 4 years - up to 22% of cases; 5-9 years - up to 41; 10-19 years - up to 85%; over 20 years of age - up to 95% of patients with neurofibromatosis type I[24]. These nodules do not occur in other forms of neurofibromatosis[37].
Iris hamartomas were first described in 1918[38]. Their importance in the diagnosis of neurofibromatosis type I was demonstrated in 1937 by the Austrian ophthalmologist Karl Lisch, after whom they received their name. Subsequently, their critical role in the differential diagnosis of Recklinghausen's disease was established[39][40].
Bone changes
Kyphoscoliosis in a patient with type I neurofibromatosis Severe
neurofibromatosis is characterized by spinal deformity in the form of scoliosis, possible marginal defects of the vertebral bodies, their articular and transverse processes, widening of the intervertebral foramina and erosion of their edges, abnormalities of the lower edges of the posterior ribs caused by pressure from neurofibromatous nodes [36] .
Long tubular bones can be atrophic, curved, and sometimes, on the contrary, hypertrophied and thickened. The compact substance in the hypertrophied bone is thickened. Periosteal ridges are visible on the surface of the bone, and sometimes paraosteal ossifications are also found. Intraosseous neurofibromas in long bones appear as localized swellings and cystic formations[36].
When the bones of the skull are involved in the process, its asymmetry is revealed. It is especially pronounced when there are deformations of the facial part and the walls of the orbit. In the bones of the cranial vault, defects and abnormalities, areas of bone atrophy or hyperostosis are possible[36].
Additional clinical manifestations of neurofibromatosis type I
- Cognitive impairment - difficulties in mastering writing, reading, and mathematics. Often combined with a moderate decrease in intelligence (IQ<70)[24]. Patients often experience depression due to shame caused by disfigurement of the body and face with neurofibromas, fear of society;
- Endocrine disorders - pheochromocytoma[41], growth and puberty disorders[24];
- Epileptic seizures[24];
- Decreased muscle tone[24];
- Behavioral disorders[24];
- Diseases not directly related to nerve involvement include stenosis of the renal and pulmonary arteries, pulmonary cysts, interstitial pneumonia, clitoral hypertrophy, and abnormal formation of the gastrointestinal tract[1]. Renal artery stenosis, especially in combination with pheochromocytoma, causes the development of arterial hypertension
- Syringomyelia[1]
MRI of the left leg: malignant tumor of the tibial nerve sheath in Recklinghausen syndrome (NF-1). Neurofibroma of the upper eyelid.
Basic diagnostic criteria
Tumors caused by radiation are different from all others
In order to make a diagnosis, at least two criteria must be present.
- 6 or more coffee stains on the child’s body. Also, the appearance of coffee stains can be associated with certain hormonal pathologies, tuberous sclerosis, Fanconi anemia.
- The presence of 2 or more neurofibromas of any type or one pleximorphic neurofibroma
- "Freckles" in the armpits and groin areas (Crove's sign)
- Lisch nodules are small, asymptomatic lesions on the iris of the eye. Lischk nodules appear in all patients with neurofibromatosis -1 by adulthood.
- Optic nerve gliomas
- Having a close relative (parent) with neurofibromatosis-1
- Characteristic bone lesion (sphenoid bone dysplasia)
The pathogenesis of neurofibromatosis is associated with imperfection of the neurofibromin gene.
Clinically, the disease is expressed in the appearance on the human skin of a certain number of formations on the stalk (from several to hundreds of thousands). Neurofibromas are pink or flesh-colored and are most often located on a stalk. The disease is often accompanied by the development of tumors in the central nervous system.
Children who have multiple coffee stains but do not meet the diagnostic criteria for neurofibromatosis should be regularly seen by an ophthalmologist (eye examination for optic nerve gliomas), pediatrician (child development assessment), orthopedic surgeon (scoliosis diagnosis), neurologist (full neurological examination) and undergo blood pressure screening.
Neurofibroma of the brain is most often a benign, relatively slow-growing formation. Stereotoxic radiosurgery is a relatively new technique for combating benign and malignant brain tumors, but, nevertheless, it is increasingly popular in the world. It can rightfully be said that, together with the discovery of radiosurgery, humanity has taken another step towards solving the mystery of how to fight cancer tumors.
It is difficult to envy people who have been diagnosed with a brain tumor - firstly, they had to accept the fact that they were seriously ill, and secondly, they had to come to terms with the prospect of brain surgery. Everyone will shudder at the thought of the inevitable craniotomy to remove the tumor. But now there is an alternative to traditional surgical treatment. Radiosurgical method for treating brain tumors, in particular. The CyberKnife installation allows you to treat such diseases bloodlessly and painlessly, and you don’t even have to go to the hospital to undergo the procedures!
In cases where doctors see the prospect of a complete recovery of the patient, they combine traditional surgical and radiosurgical methods, which complement each other. For example, in clinics equipped with a “Cyber Knife” the following scheme is often practiced: surgical removal of the tumor, preventive irradiation of the tumor bed, MRI examination once every three months, so as not to miss a relapse—the beginning of a new tumor. If the formation does appear again, it is irradiated to “freeze” its growth and prevent it from increasing in size. This scheme is highly effective and sometimes really allows you to snatch the patient from the “clutches of death.”
"Cyber Knife" is indispensable in cases where a brain tumor:
- located in a place difficult to reach for a surgeon’s scalpel
- adjacent to the most important functional parts of the brain
- represented by small superficially located multiple formations
Traditional surgery is abandoned in such cases, since the risk of complications is too great. The most promising for surgical removal and prognostically favorable are slowly growing benign formations that do not “invade” their cells into neighboring healthy tissues and have clear, well-differentiated edges. Such tumors are dangerous only due to compression of brain structures, which is much less dangerous than the activity of a malignant tumor, which destroys its tissue.
(495) 740-58-05 – information on the Cyber Knife system
Notes
- ↑ Kozlov A.V.
Neurofibromatosis 1 (NF1) // Surgery of skull base tumors / Edited by A. N. Konovalov. - M.:: OJSC "Mozhaisk Printing Plant", 2004. - P. 166-169. — 372 p. — 1000 copies. — ISBN 5-94982-006-1. - . Retrieved August 16, 2010.
- Schneider N.
A. website krasmedic.ru. Retrieved May 15, 2011. - (unavailable link). website doctoram.net. Retrieved May 15, 2011.
- . website myemergency.ru. Retrieved May 13, 2011.
- . website www.medfix.ru. Retrieved May 13, 2011.
- (English). BBC News (21-07-2003). Retrieved May 13, 2011.
- Roger Highfield.
(English). The Telegraph (13-05-2003). Retrieved May 13, 2011. - ↑
Schneider N.A. website www.krasmedic.ru. Retrieved May 6, 2011. - M. Deckert, G. Reifenberger, U.-N.
Riede, W. Schlote, D. R. Thal, O. D. Wiestler. Nervensystem // Allgemeine und Spezielle Pathologie / Ursus-Nikolaus Riede, Martin Werner, Hans-Eckart Schäfer. - 5. - Stuttgart, 2004. - P. 1104. - (unavailable link). website "Knowledge Base on Human Biology". Retrieved May 7, 2011.
- Kluwe L., Tatgiba M., Fünsterer C., Mautner V.-F.
[www.ncbi.nlm.nih.gov/pmc/articles/PMC1735482/pdf/v040p00368.pdf NF1 mutations and clinical spectrum in patients with spinal neurofibromas] // J Med Genet. - 2003. - T. 40. - P. 368-371. - ↑
- ↑
- Doran SE, Thorell WE
Chapter 40. Brain Tumors // Youmans Neurological surgery / Winn HR. — 5th edition. - Philadelphia: Saunders, 2004. - Vol. 1. - P. 812. - ISBN 0-7216-8291-x. - Solovyov Yu. N.
Neurofibroma // Great Medical Encyclopedia / under the general editorship of B. V. Petrovsky. — 3rd edition. - M.:: “Soviet Encyclopedia”, 1981. - T. 16. - P. 309-310. — 512 s. — 150,000 copies. - ↑ Özek MM, Urgun K.
Tumors of the Optic Pathway and Hypothalamus // Essential Practice of Neurosurgery / Editor-in-Chief Kazadi KN Kalangu, Deputy Editors Yoko Kato and Gilbert Dechambenoit. - Nagoya, Japan: Yamagiku Printing Co., Ltd. — P. 348. — 1496 p. — ISBN 978-4-904992-01-2. - Davis FA
Primary tumors of the optic nerve (a phenomenon of Recklinghausen`s disease) (English) // Arch Ophth. - 1940. - Vol. 23. - P. 957-1018. - Bondy M., Wrensch M., Wiencke J.
Genetic and hereditary syndromes associated with tumors of the CNS // Cancer in the Nervous System / Levin. — 5th edition. - New York: Churchill Livingstone, 1996. - P. 1-12. - ↑ Melnikov R. A., Kitaev V. V.
Neurofibromatosis // Great Medical Encyclopedia / under the general editorship of B. V. Petrovsky. — 3rd edition. - M.:: “Soviet Encyclopedia”, 1981. - T. 16. - P. 310-311. — 512 s. — 150,000 copies. - ↑. website lechenee.ru. Retrieved May 7, 2011. (inaccessible link)
- PJ Waardenburg.
Heterochrome en melanosis // Nederlands tijdschrift voor geneeskunde. - Haarlem, 1918. - T. 62. - P. 1453-1455. - Symptoms and syndromes in endocrinology / Ed. Yu. I. Karachentseva. — 1st ed. - Kh.: LLC "S.A.M.", Kharkov, 2006. - P. 141. - 227 p. — (Reference manual). — 1000 copies. — ISBN 978-966-8591-14-3.
- Neurofibromatosis.
Conference Statement, NIH Consensus Development Conference.
in: Archives of neurology.
official organ of the American Neurological Association. Chicago Ill 45.1988, 575—578 - . website emed.nextday.su. Retrieved May 7, 2011. (inaccessible link)
- Kozlov A. V.
Neurofibromatosis 1 (NF1) // Surgery of skull base tumors / Edited by A. N. Konovalov. - M.:: OJSC "Mozhaisk Printing Plant", 2004. - P. 169-170. — 372 p. — 1000 copies. — ISBN 5-94982-006-1.
How does neurofibroma manifest?
Neurofibroma grows slowly and rarely manifests clinical symptoms. As the formation reaches a large size, the patient develops neurological symptoms. Typically, neurofibroma is a solitary tumor with well-differentiated margins. With timely treatment, the prognosis is favorable.
A disease that deserves special attention is multiple neurofibromatosis (Recklinghausen's disease), a hereditary pathology that belongs to the group of dysplastic processes. Neurofibromatosis is divided into 2 types, the 1st type of this disease is much more common. If we talk about the prevalence of neurofibromatosis -1, then according to medical statistics, it affects 1 person out of 3000.
General information
Neurofibromas are benign tumors that develop from the sheaths of nerve fibers. Most often they are located in the layers of the skin and subcutaneous tissue, sometimes affecting the brain, nerve fibers, spinal cord roots, soft tissues, and internal organs. Neurofibromatosis is a disease in which numerous neurofibromas are formed.
The prevalence of different types of pathology varies significantly: the incidence of type 1 is 1:2,500, type 2 – 1:50,000. Other variants are even less common, their exact epidemiology has not been determined. No gender or racial predisposition was identified. The onset of clinical manifestations is possible at any age, depending on the type of disease.
Neurofibromatosis
Neurofibromatosis type 2
Diagnosis of neurofibromatosis 2 may be more difficult when family history is not available. Hearing loss that begins in the teens or early twenties is often the first symptom. People with this disease usually develop tumors of the auditory nerve or suffer from other related complications.
Auditory nerve tumor
Most people affected by neurofibromatosis-2 have tumors of the nerves that perform auditory function (acoustic neuroma, acoustic schwannoma, acoustic neuroma, auditory neuroma, vestibular schwannoma). Although tumors are usually benign, they often lead to progressive hearing loss due to their gradual increase in size.
Other complications
People with neurofibromatosis 2 also experience ringing in the ears, headaches, facial pain/numbness, and vestibular problems, among other symptoms.
Diagnostic methods
The content of the pictures suggests that the diagnosis of neurofibromatosis is characterized by an extremely thorough approach. The suspected patient is prescribed an MRI and CT scan of the spine and brain, an X-ray of the skull and spine, hearing and vision tests are performed, and consultation with a neurologist, orthopedist, or neurosurgeon is required.
Treatment of neurofibromatosis
Treatment of neurofibromatosis
The Israeli Center for Neurosurgery and Neurology Neuromed specializes in the conservative and surgical treatment of neurofibromatosis using innovative techniques to increase the effectiveness and safety of treatment.
What is neurofibromatosis
Neurofibromatosis is a group of three genetically distinct disorders that cause tumors to grow in the nervous system. These tumors arise in the supporting cells that make up the nerve and the myelin sheath (the thin membrane that envelops and protects nerves). Scientists classify neurofibromatosis into types 1 and 2. Most tumors are benign, but some can become malignant.
The reasons for the development of this pathology are not reliably known, but it is believed that its occurrence is associated with genetic mutations that play a key role in suppressing cell growth in the nervous system.
Neurofibromatosis type 1
Neurofibromatosis type 1 is the most common type of this pathology and occurs on average in 1 person in 4000. Most people inherit the disease, but about 30% of new cases of the pathology appear as a result of a genetic mutation that occurs for unknown reasons. Once a disease appears as a result of mutation, the mutated gene can be passed on to subsequent generations.
Signs of neurofibromatosis type 1
To diagnose neurofibromatosis, the doctor looks for the presence of at least two of the following signs:
- six or more light brown spots on the skin, more than 5 mm in diameter in children and more than 15 mm in diameter in adolescents and adults;
- two or more neurofibromas;
- freckles in the armpits or groin area;
- two or more growths on the iris of the eyes;
- tumor of the optic nerve (glioma);
- developmental anomalies of the spine (scoliosis), sphenoid cranial bone, pathology of the tibia;
- parent, sibling or child has neurobiphromatosis type 1.
Many children with neurofibromatosis have a larger head circumference than normal. Hydrocephalus (abnormal accumulation of fluid) in the brain is a possible complication of the disease. People with neurofibromatosis type 1 have seizures and frequent headaches, as well as cardiovascular disease, congenital heart defects, high blood pressure (hypertension), and narrowed, blocked, or damaged blood vessels (vasculopathy).
Children with this disease have poor speech and impaired visual-spatial skills, as well as some educational delays. In addition, a sign of neurofibromatosis in children is mental retardation, attention deficit disorder and/or hyperactivity.
The most common signs of neurofibromatosis, such as skin spots, neurofibromas, freckles in the armpit and groin, are visualized immediately after birth or after some time. Many features of these signs depend on age, so a final diagnosis may take several years.
Treatment of neurofibromatosis type 1
Surgery is recommended for the treatment of neurofibromatosis that can become malignant, as well as for tumors that cause significant visual defect. There are several options for surgical treatment, but among doctors there is no common vision of the progress of the operation in one way or another. Treatment for neurofibromas that have become cancerous may include surgery, radiation, and chemotherapy. These methods can also be used to reduce the size of optic nerve gliomas and control the disease. Some bone abnormalities, such as scoliosis, can be corrected with surgery.
Treatment for other conditions associated with neurofibromatosis is aimed at managing or relieving symptoms. Headaches and seizures are treated with medications. Children with this pathology are at high risk of disability, so they should undergo a thorough neurological examination before entering school, as well as tests to assess verbal and spatial skills.
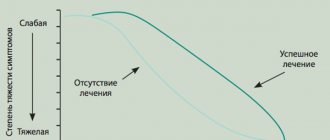
Forecast
Neurofibromatosis is a progressive disease, so symptoms will worsen over time. Only in some people the signs of the disease remain unchanged for a long time. The course of the disease for an individual patient is difficult to predict, but most people experience mild or moderate symptoms over time and have a normal life expectancy. A neurofibroma under or on the skin may grow larger over time.
What is the prognosis for the disease?
There are no reliable prognostic criteria that could predict the course of the disease for neurofibromatosis. In neurofibromatosis type 1, life expectancy is shortened only with the early development of plexiform neurofibromas.
For all other patients, life expectancy, as a rule, does not differ from the average life expectancy in the population. The presence of neurofibromatosis type 2 also does not have a significant effect on life expectancy. The growth rate of vestibular schwannomas is usually slow. In rare cases, their rapid increase occurs.
Patients with neurofibromatosis type 2 are not recommended to dive or swim without safety nets due to the high risk of drowning.
Treatment code
Treatment is surgical. Indications for it are severe pain or ulceration of the tumor, difficulty moving, compression or displacement of vital organs. In some cases, surgery is performed for cosmetic purposes. Since the lesions in neurofibromatosis are multiple, removal of all pathological foci, in most cases, is not possible.
Surgical treatment of the elephantine form of neurofibromatosis requires subsequent skin grafting. The tissue of neurofibromas is abundantly supplied with blood vessels. When the node is located in a large nerve trunk, the tumor is enucleated, the nerve is resected with the application of a nerve suture, or its marginal resection is performed with a partial nerve suture is applied. Surgical removal of one of the nodes in some cases can lead to progression of the process with a sharp increase in the size of other nodes.
As of the first half of 2011, pathogenetic treatment (that is, aimed at the main mechanisms of disease development) is in phases I-II of clinical trials and is not widely used. There is evidence of the effectiveness of Ras inhibitors (tipifarnib) in the treatment of neurofibromatosis type I. Pirfenidone has also been shown to be effective in animals. However, until clinical trials are completed, these and a number of other drugs cannot be used in the treatment of neurofibromatosis.
What is Neurofibromatosis, Recklinghausen's disease?
This disease is usually classified as a special group of pathologies - phakomatoses.
They represent metabolic disorders in the body as a whole, associated with damage to the gene encoding the synthesis of one or another important enzyme. As a result, many organs and systems are affected, mainly malformations of the eyes, skin, nervous system, and a number of internal organs are observed.
So, we have determined that neurofibromatosis is a phakomatosis with a predominant lesion of the skin.
The study of this pathology is a big problem. In particular, it cannot be attributed to one or another section of medicine, since, in fact, it relates a little to each of them. Damage to multiple organs greatly complicates treatment. In addition, the disease can ultimately lead to the development of various skin tumors, and the mechanism of this phenomenon is also almost completely unknown, which means that appropriate prevention methods have not been developed.
Pathogenesis (what happens?) during Neurofibromatosis (Recklinghausen's disease):
Pathology manifests itself in the form of proliferation in all organs and tissues of benign tumors, which consist mainly of connective tissue and pigment cells (on the skin). Tumors are formed around the nerve trunks, growing from their membranes - neuromas or neurofibromas. Mostly pigment spots and formations characteristic of liver damage (fatty plaques, dilated vessels in the form of cysts) appear on the skin. This is due to damage to the liver tissue by tumor cells. Formations in the form of pigment spots also appear on the retina of the eyes, as a result of which the patient’s vision is greatly affected. This phenomenon is called retinal phakomatosis. On the oral mucosa, on the contrary, tumors mainly form, consisting of connective tissue, which look like small elastic dense nodules
They can be large in size; their flat and smooth surface always attracts attention.
In most cases, they rise above the level of the mucous membrane, sometimes they can be located deep in the tissue, which makes them difficult to detect. Great diversity is noted in terms of changes in the skeleton of the torso, arms and legs. The bones may be poorly developed or, on the contrary, be significantly thickened, giving the limb an ugly shape, disrupting the patient’s normal life. The most permanent and persistent change is a significant curvature of the spinal column, which severely impairs the functions of the spinal cord.
On the part of the nervous system, such children, as a rule, exhibit one or another degree of decline in intellectual development; they often fall into a state of severe depression, from which it is sometimes difficult to bring the patient out of it. The pathological gene is inherited as an autosomal dominant trait, so all children who receive it are sure to get sick. There can be no carrier state. With the greatest frequency, the pathology affects males, but no connection has been established between the inheritance of neurofibromatosis and gender. Women suffer about half as often.
Treatment of Neurofibromatosis (Recklinghausen's disease):
Just as with ichthyosis, there are no radical methods of therapy. Drugs are used that promote some normalization of disturbed metabolic processes. The method and course of treatment is selected for each patient purely individually with the participation of a geneticist, neurologist, ophthalmologist, orthopedist, oncologist, etc. The only method of treating cosmetic defects caused by tumor processes is surgery. Therefore, a pediatric surgeon is also involved in the treatment of such patients.
New generations of medications are recommended, the action of which is aimed at improving metabolism in connective tissue, the nervous system, and internal organs.
Forecast
The prognosis for life is favorable. In terms of ability to work, it is favorable if there is no pathology of the skeletal system and mental retardation is slightly expressed.
Rehabilitation
Rehabilitation measures for neurofibromatosis are built in accordance with the severity of the disease, the presence of lesions of the skeletal system (limitation of physical activity, classes with an orthopedist, therapeutic physical education) and the nervous system (training in special schools, classes with a psychologist, neurologist). Since the disease is lifelong, such patients must be constantly monitored by appropriate specialists throughout their lives.
Story
The first scientific description of the clinical and morphological changes of this disease was made in 1882 by the German pathologist Friedrich von Recklinghausen. The description of the disease was made long before the discovery of the structure of DNA. In this regard, it was called “Recklinghausen’s disease”. This term denoted not only neurofibromatosis type I, but also neurofibromatosis in general. Neurofibromatosis types I and II, different in their causes and some manifestations, were defined as “peripheral” and “central” forms of Recklinghausen’s disease. After determining the genetic causes of the occurrence, the term began to become obsolete. In modern medical literature, the disease is called “neurofibromatosis type I.”
It was long believed that Joseph Merrick, known for his deformities, nicknamed the “Elephant Man,” had neurofibromatosis type I. David Lynch's famous film "The Elephant Man" about the plight of John Merrick contributed to the emergence in society of a false opinion that people with this disease have a terrible appearance. Modern researchers suggest that Merrick has Proteus syndrome.
Epidemiology
Autosomal dominant type of inheritance
The incidence of neurofibromatosis type I is 30-40 patients per 100 thousand population, which corresponds to one person in 2500-3000. Neurofibromatosis type I is an autosomal dominant disease with high penetrance and a high incidence of new mutations. The risk of having a child with neurofibromatosis from a sick person is either 50% (in the case of a heterozygote) or 100% (in the case of a homozygote). Approximately 50% of cases of the disease are mutations of lat. de novo[11]. The incidence of neurofibromatosis type I does not vary across geographic regions or ethnic groups[12].
Neurofibromatosis causes, diagnosis, treatment
One of the more common diseases among hereditary diseases is neurofibromatosis. Few people know what this is, since in general the pathology is observed quite rarely.
Considering that neurofibromatosis (NF) has two types, the first type (peripheral) is diagnosed in one case per 3500-4000 newborns, and the second (central) is even less common - one case among 50,000 newborns.
The disease is a tumor in the nerve endings containing various elements, including fibroblasts, mast cells and Schwann cells. To fully understand what neurofibromatosis is, it is worth familiarizing yourself in more detail with general information, symptoms and methods of treatment.
What is neurofibromatosis
In medicine, neurofibromatosis is also called Recklinghausen's disease, and belongs to the group of autosomal dominant diseases. Neurofibromatosis is caused by a sudden mutation of a certain gene, resulting in the formation of tumors.
Neurofibromas are benign tumors that form along the trunks of nerves and their branches. Most often, they begin to develop in children after ten years of age, but sometimes neurofibromatosis can also occur in adults, localized in the skin or subcutaneous tissue.
In some cases there may be integrated development in both areas.
Most often, the main factor due to which Recklinghausen syndrome develops is genetics, when one of the parents had this disease or the presence of relatives, but the disease can also develop with the formation of other mutations.
Causes
Causes of development of Recklinghausen neurofibromatosis
Recklinghausen's neurofibromatosis can be diagnosed in two ways:
- Heredity - this disease can be transmitted from parents to children, the probability of this is 50%. If even one defect is found in the DNA, the disease will develop.
- Unexpected gene mutation - there are cases when no one in the family suffered from neurofibromatosis, but due to certain genetic failures it still appeared. The NF-1 gene is responsible for the formation of tumors and the development of Recklinghausen's disease.
In addition, hormonal changes, such as puberty, pregnancy, diseases of the endocrine system, injury and taking certain medications, can accelerate the development of cells that form neurofibromas.