Синдром Эдвардса является одной из форм редкого генетического заболевания, когда часть 18-хромосомы человека дублируется. Большинство детей с данной патологией умирают еще на стадии эмбрионального развития, это происходит в 60 % случаев. Распространенность синдрома Эдвардса в среднем составляет 1:3000—1:8000 случаев. Наследование синдрома не прослеживается, а случайность данной мутации составляет всего 1%.
Синдром Эдвардса был назван в честь доктора Джона Эдварда, который в 1960 году описал первые случаи и зафиксировал закономерность развития симптомов. Синдром Эдвардса затрагивает больше женский пол, чем мужской — около 80 процентов пострадавших составляют женщины. Женщины старше тридцати лет имеют больший риск рождения ребенка с синдромом, хотя то же может происходить и с женщинами моложе тридцати, но значительно реже. Около 12% детей с синдромом доживают до возраста, когда можно оценить возможности умственного развития; младенцы, которые выживают, имеют серьезные дефекты и обычно живут не долго. Синдром Эдвардса связан с широким спектром нарушений, которые состоят более чем из ста тридцати дискретных дефектов, связанных с мозгом, сердцем, черепно-лицевой структурой, почками и желудком.
Причины развития синдрома Эдвардса.
Каждая клетка в организме человека содержит в себе двадцать три пары хромосом, которые он наследует от родителей. У людей в каждой половой клетке содержится одинаковое число хромосомных наборов. У женщин это яйцеклетки (их называют XX), а у мужчин (их называют XY) это сперматозоиды. Во время деления оплодотворённой яйцеклетки происходит мутация под воздействием каких-либо факторов и возникает дополнительная хромосома в восемнадцатой паре, которая и несет ответственность за возникновение синдрома Эдвардса. Дети с синдромом наследуют неправильное число хромосом, вместо двух копий у них образуется три копии хромосом. Данный вид мутации называют «трисомия», так же к названию прилагается номер пары, в которой произошла мутация, в случае синдрома Эдвардса это восемнадцатая пара.
Выше описанный вариант является «полной трисомией», т. е. ребенок унаследовал полную дополнительную копию лишней хромосомы, таких детей с синдромом Эдвардса по статистике 95%. «Полная трисомия» имеет практически все признаки заболевания и протекает очень тяжело.
Существует еще два варианта мутаций. Два процента детей с синдромом Эдвардса имеют транслокацию в 18 паре, когда присутствует только часть лишней хромосомы. Три процента детей синдромом имеют «мозаичную трисомию», данный вид мутации характеризуется наличием лишней хромосомы не во всех клетках организма.
Причины возникновения
Как указывалось выше, заболевание генетическое, в его основе лежит мутация – появление третьей (дополнительной) 18 хромосомы. В результате этого в генетическом наборе присутствует не 46, а 47 хромосом.
Причины генетической мутации не выяснены. Считается, что факторы риска следующие – возраст матери, ее вредные привычки и наличие хронических заболеваний, плохие условия жизни и работы (постоянное воздействие неблагоприятных факторов окружающей среды). Также значение имеет отягощенная наследственность матери ребенка.
Симптомы синдрома Эдвардса.
Большинство детей, рожденных с синдромом Эдвардса, имеют дефицит массы тела и выраженную задержку в развитии. Их голова необычно мала, а затылок имеет выраженный размер. Их уши низко посажены, верхняя и/или нижняя челюсть имеет дефект развития, называемый микрогнатией. Это состояние, когда искажается форма лица и формируется неправильный прикус.
Внешний вид больного с Синдромом Эдвардса
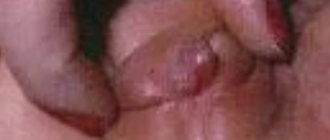
У детей часто формируется «волчья пасть» и «заячья губа», состояния, когда возникает расщелина в верхнем небе и губе соответственно.
Сформировавшаяся «заячья губа»
Ручки детей часто сжаты в кулачки и все пальцы имеют характерную неровную позицию.
Флексорное положение кисти
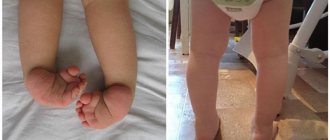
Дети страдают косолапостью, а пальцы на нижних конечностях могут иметь перепонки или полностью сливаться между собой.
Очень часто у больных с синдромом возникают проблемы с сердцем, легкими и диафрагмой из-за патологии сосудистого звена и развития врожденных пороков. Сердечные пороки могут иметь различный характер и различное количество: открытый артериальный проток, дефект межжелудочковой перегородки, открытое овальное отверстие.
Мутации могут вызывать развитие паховых и пупочных грыж, аномалии развития мочеполовой системы и диспластический синдром. Процентное соотношение развития того или иного порока:
Поражение системы и порок (признак) — Частота %Мозговой череп и лицо — 100% микрогения — 96,8% низкое расположение или/и деформация ушных раковин — 95,6% долихоцефалия — 89,8% высокое небо — 78% расщепленное небо — 15% микростомия — 71% Опорно-двигательный аппарат — 98,1% флексорное положение кистей — 91,4% стопа-качалка — 76,2% кожная синдактилия стоп — 49,5% косолапость — 34,9% Центральная нервная система — 20,4% Сердечно-сосудистая система — 90,4% Органы пищеварения — 54,9% Мочеполовая система — 33,5%
У детей с синдромом Эдварда, как правило, возникают проблемы с кормлением. Проблемы возникают из-за нарушения врожденных рефлексов глотания и сосания. Слабое развитие сосательного рефлекса и несогласованное глотание приводит к удушью ребенка.
Ребенок может заболеть гастроэозофагиальной рефлюксной болезнью. Врожденные расщелины лицевого скелета также вызывают трудности в скармливании. Они приводят к необходимости кормить ребенка через зонд или гастростому. Специалист может показать родителям ребенка, как правильно держать голову и тело своего ребенка. В целях предотвращения рефлюкса голова ребенка должна быть приподнята примерно на тридцать градусов или более пока ребенок ест, и в течение часа или двух после того как он поел.
Формы синдрома
На тип такой аномалии в первую очередь влияет стадия развития зародыша, на которой синдром настигает эмбрион. Всего различаются три вида:
- Полный. Самый тяжелый тип, на него приходится 80 % случаев. Утроенная хромосома появляется в тот момент, когда плод был всего одной клеткой. Отсюда следует, что аномальный хромосомный набор будет передаваться при делении и всем остальным клеткам, наблюдаться в каждой из них.
- Мозаичный. Название дано из-за того, что здоровые и мутировавшие клетки смешаны, как мозаика. 10 % пораженных симптомом Эдвардса страдают именно этой формой. Признаки болезни здесь выявлены слабее, но все же мешают нормальному развитию ребенка. Избыточная хромосома появляется во время фазы, когда зародыш состоит из нескольких клеток, поэтому поражается только часть организма или отдельный орган.
- Возможная транслокация. Здесь наблюдается не только нерасхождение хромосом, но и переизбыток информации, порожденный транслокационной перестройкой. Проявляется как во время созревания гамет, так и во время развития зародыша. Отклонения здесь не ярко выражены.
Диагностика синдрома Эдварда
Диагноз синдрома Эдвардса может быть поставлен путем определения при физическом осмотре всех вышеперечисленных внешних симптомов. Более точный диагноз можно поставить, проведя анализ «кариотипирования», который включает в себя взятие образцов крови ребенка для определения хромосомного состава.
В связи с большим количеством пороков развития и очень низким процентом выживаемости детей с синдромом Эдвардса, в настоящее время разработаны методы антенатальной диагностики. Одной из первых и доступных методик, которые осуществляют во всех женских консультациях, является УЗИ плода. На УЗИ в ранних сроках беременности можно заподозрить пороки развития головного мозга и конечностей, также наличие обильного количество околоплодной жидкости, что должно насторожить врача. При получении данных результатов необходимо отправить беременную на более детальное и прицельное наблюдение в стационар.
В стационаре должны выполнить все необходимые общеклинические анализы и провести более специфические тесты, такие как эхография, доплерометрия, исследование сывороточных маркеров крови: β-субъединицы хорионического гонадотропина (βХГ), α-фетопротеина (АФП), эстриола (ЕЗ), 17-окси прогестерона. Для оценки степени риска рождения детей с хромосомной патологией во всех наблюдениях используют специально разработанную компьютерную программу PRISCA, учитывающую возраст женщины, показатели сывороточных маркеров и срок беременности. Также необходимо выполнить трансабдоминальный амниоцентез с последующим кордоцентезом. В амниотических водах при наличии синдрома Эдвардса будет определяться: АФП, 17-ОП, ЕЗ, а в крови плода будет определяться кариотип трисомии 18 хромосомы.
Диагностика
Синдром Эдвардса является показанием для прерывания беременности, поэтому его наличие необходимо выявить как можно скорее. Сделать это можно при помощи пренатального скрининга – комплекса лабораторных исследований материнской сыворотки на гормоны, в том числе и те, которые вырабатывает плодная оболочка:
- бета-ХГЧ;
- РАРР-тест;
- АФП (альфа-фетопротеин);
- свободный эстриол.
Заподозрить наличие у ребенка данной патологии можно также при проведении УЗИ и допплерографии маточно-плацентарного кровотока. Конечно, результаты таких исследований могут быть косвенными, и не являться окончательным «вердиктом». Это касается как инструментальных, так и лабораторных методов диагностики.
Оценка риска рождения ребенка с таким отклонениям проводится с учетом:
- срока беременности;
- возраста будущей матери;
- данных биохимического и ультразвукового скрининга;
- массы тела беременной женщины.
Беременным, относящимся к группе высокого риска, врачи предлагают пройти дородовую диагностику, заключающуюся в проведении биопсии хориона, амниоцентеза, кордоцентеза. После этого выполняется кариотипирование плода.
Если синдром Эдвардса был выявлен у ребенка после его рождения, необходимо незамедлительно провести всестороннее обследование для выявления тяжелых пороков развития. Для этого осуществляется осмотр целым рядом специалистов:
- кардиологом;
- неврологом;
- педиатром;
- хирургом;
- ортопедом;
- урологом.
В первые часы после рождения ребенка с предполагаемым диагнозом «синдром Эдвардса» важно провести ЭхоКГ, УЗИ органов брюшной полости и почек.
Лечение синдрома Эдвардса
Научному медицинскому сообществу не известно лекарство от синдрома в настоящее время. Дети с синдромом Эдвардса обычно рождаются с физическими отклонениями, и врачи сталкиваются с проблемой в отношении выбора метода лечения. Хирургия может устранить некоторые пороки, связанные с синдромом, но инвазивность методики не оправдывает результата у детей, чей срок жизни колеблется от нескольких дней до месяцев.
Лечение на сегодняшний день сводится к паллиативной помощи (поддержанию морального духа столкнувшихся со смертельным заболеванием). От пяти до десяти процентов детей с синдромом Эдвардса доживают до первого года жизни.
Проблемы, связанные с нарушениями нервной системы и тонуса мышц влияют на развитие двигательных навыков ребенка, что может привести к сколиозу, атрофии мышц, косоглазию. Хирургическое лечение у больных детей может быть ограничено из-за пороков развития сердечно-сосудистой системы. У детей с синдромом часто возникают запоры, вызванные плохим тонусом брюшной стенки и атоничным кишечником. В результате этого дети грудного возраста испытывают дискомфорт, возникают трудности с кормлением. Специальные молочные смеси, слабительные средства и препараты из группы «пеногасителей» могут облегчить данные симптомы. Клизмы в данной ситуации противопоказаны, потому что они могут привести к электролитному дисбалансу у ребенка.
Дети с синдромом часто отстают в развитие, в связи с этим необходимо применение специально разработанных программ терапии по коррекции задержки физического развития.
Еще одним фактором неблагоприятного прогноза является повышенный риск развития опухоли «Вильмса» – это одна из разновидностей рака почки. Рекомендуется регулярное УЗИ обследование брюшной полости на наличие данной патологии.
Больные дети подвержены риску развития различных инфекций и патологических состояний, таких как: инфекции мочеполовой системы, средний отит, конъюнктивиты различной этиологии, синуситы, фронтиты, пневмонии, ночное апноэ, легочная гипертензия, врожденные пороки сердца и повышенное артериальное давление. Необходимо быть готовыми, чтобы эффективно провести лечение и вовремя заподозрить данные заболевания.
Родителям, у которых ребенок имеет данный синдром, необходимо постоянное наблюдение за состоянием здоровья, так как своевременно выявленное заболевание может продлить жизнь их детям.
Что делать родителям, если обнаружен Синдром Эдвардса у плода
Точные причины хромосомной мутации не выявлены до сих пор. Есть риск рождения деток с патологиями у возрастной матери, но в случае с данным Синдромом больной ребенок может родиться у фактически здоровых мамы и папы. Главное, что могут сделать родители – это пройти все обследования на этапе планирования беременности, если нужно, получить консультацию у генетика.
Все симптомы болезни можно обнаружить на УЗИ, которое проводится в конце первого триместра беременности.
Самая сложная часть такой проблемной беременность – принять решение. Синдром Эдвардса является значительно более тяжелой патологией, чем Синдром Дауна. В последнем случаи эти детки могут преуспеть в жизни, найти себя, быть социально полезными. Синдром Эдвардса фактически не оставляет шансов для этого.
Потому при пренатальном обнаружении патологии:
- Родители в большинстве случаев решаются на прерывание беременности, которое в варианте с этим диагнозом возможно и на более позднем сроке, если патология была обнаружена не сразу;
- После прерывания беременности родители проходят консультации генетиков для выявления возможных причин таких нарушений.
Рождение таких больных деток в редких случаях является осознанным выбором родителей. Чаще это дети матерей, которые не становились на учет по беременности, и не проходили всех возможных исследований. Обычно родители отказываются от ребенка, практически обреченного на гибель.
Стоит знать, что не увидеть патологию на УЗИ почти невозможно. Потому так важно беременной в период гестации соблюдать все предписания врача, сдавать вовремя все анализы, в полном объеме пройти первый скрининг. Если благодаря исследованиям будут обнаружены тяжелые патологии, прерывание беременности в первом триместре будет не так болезненно, не так психологически травматично для матери, как поздний аборт или искусственные роды. Почти в любом медучреждении сегодня работает психолог, к которому может обратиться женщина (или оба родителя), оказавшиеся в столь сложной ситуации.
Прогноз синдрома Эдварда
Прогноз в большей своей степени при данной патологии не благоприятный. Большинство детей, которые родились с синдромом Эдвардса, не доживают до первого года своей жизни. Средняя продолжительность жизни для половины детей, рожденных с этим синдромом, менее чем два месяца. От девяноста до девяноста пяти процентов из этих детей умирает, не дожив до своего первого дня рождения. От пяти до десяти процентов детей, которые выжили в первый год, испытывают серьезные отклонениями в развитии.
Дети, которые прожили первый год, требуют постоянного контроля и наблюдения, и испытывают серьезные трудности в развитии. Их словесные навыки общения ограничены, хотя они в состоянии реагировать на утешения своих родителей и имеют возможность научиться улыбаться, распознавать и взаимодействовать с воспитателями и другими людьми. Они могут приобрести навыки, необходимые для всех детей, такие как самостоятельное кормление и поднятие головки, навыки, характерные для здоровых детей их возраста.
Врач терапевт Жумагазиев Е.Н.
Причины
К сожалению, причины мутации врачами до сих пор не выяснены. Известно лишь, что девочки подвержены такой патологии в несколько раз чаще, чем мальчики, и что у мам в возрасте за 30 вероятность рождения малыша с таким отклонением, как синдром Эдвардса повышается.
Причины появления синдрома Эдвардса у ребёнка не зависят от состояния здоровья родителей – мать и отец могут быть полностью здоровы, как и их ближайшие родственники, но ребёнок может родиться с патологией. А так как причины установить нельзя, значит, нельзя и предотвратить развитие болезни у плода. Очень важно чтобы каждая беременная своевременно становилась на учёт и обследовалась, что позволяет выявить у её малыша синдром Эдвардса на ранних сроках, и прервать беременность.
Иногда родители решают сохранить жизнь больному ребёнку – в этом случае важно понимать, что если ребёнок не умрёт ещё в младенчестве, ему всю жизнь будет требоваться посторонняя помощь и никакое лечение не сможет ему помощь.
Отметим, что смертность в утробе матери таких детей составляет 60%. Среди тех, кто все же родился с отклонением, на первом году жизни умирают в 90% случаев, и лишь единицы доживают до подросткового или более старшего возраста. То есть прогноз течения такой патологии крайне неблагоприятен.
Дородовая диагностика
Синдром Эдвардса на УЗИ возможно определить только по косвенным признакам. Самый точный метод диагностики синдрома у плода сегодня — перинатальный скрининг. На его основе при возникновении тревожных подозрений доктор уже направляет женщину на инвазивное тестирование.
Скрининг, выявляющий кариотип синдрома Эдвардса, разделяется на два этапа:
- Первый проводится на 11-13 неделе беременности. Исследуются биохимические показатели — проверяется кровь матери на уровень гормонов. Результаты на этом этапе не окончательны — они могут говорить только о наличии риска. Для расчетов специалисту нужен белок А, ХГЧ, белок, вырабатываемый оболочками эмбриона и плаценты.
- Второй этап уже направлен на точный результат. Для исследований берется образец пуповинной крови или околоплодной жидкости, который затем повергается генетическому анализу.
Инвазивное тестирование
Хромосомы синдрома Эдвардса вероятнее всего определить данным методом. Однако он обязательно предполагает оперативное вмешательство и проникновение в оболочки эмбриона. Отсюда риск прерывания беременности и развития осложнений, отчего тест назначается только в крайних случаях.
На сегодня известны три типа взятия образца:
- БВХ (биопсия ворсин хориона). Основное преимущество метода — образец берется, начиная с 8-й недели беременности, что позволяет на ранних сроках определить осложнения. Для исследований нужен образец хориона (один из слоев оболочки плаценты), структура которого схожа со структурой эмбриона. Данный материал позволяет диагностировать внутриутробные инфекции, генетические и хромосомные болезни.
- Амниоцентез. Анализ проводится, начиная с 14-й недели беременности. В этом случае зондом протыкаются амниотические оболочки эмбриона, инструмент собирает образец околоплодных вод, содержащих в себе клетки будущего ребенка. Риск развития осложнений от такого исследования гораздо выше, чем в предыдущем случае.
- Кордоцентез. Срок — не ранее 20-й недели. Здесь берется образец пуповинной крови эмбриона. Сложность в том, что при взятии материала у специалиста нет права на ошибку — он должен попасть иглой точно в сосуд пуповины. На практике это происходит так: через переднюю стенку брюшины женщины вводится пункционная игла, которая собирает порядка 5 мл крови. Процедура проходит под контролем УЗИ-устройств.
Все вышеописанные методы нельзя назвать безболезненными и безопасными. Поэтому их проводят только в случаях, когда риск генетического заболевания у плода выше риска развития осложнений от взятия материала для анализа.
Родителям надо помнить, что ошибка медика при процедуре может привести к проявлению серьезных заболеваний, врожденных пороков у будущего ребенка. Нельзя исключить и риск внезапного прерывания беременности от подобного вмешательства.