Of the various morphological variants of soft tissue sarcomas, the most common is rhabdomyosarcoma (RMS)
, which, according to the unanimous opinion of numerous authors, accounts for about half of soft tissue sarcomas in children.
According to the Research Institute of Pediatric Oncology and Hematology, Russian Cancer Research Center named after. N. N. Blokhin RAMS (Research Institute of Children's Education), RMS accounts for about 38% of soft tissue sarcomas in children, angiogenic malignant tumors (about 22%) and synovial (19%) are less common. Other types of soft tissue neoplasms, such as leiomyosarcoma, fibrosarcoma, liposarcoma, etc., are diagnosed much less frequently.
Rhabdomyosarcoma develops from striated muscle or embryonic rudiments of muscle tissue. It consists of myoblasts at different stages of cell differentiation, in the cytoplasm of which myofibrils are found.
A certain number of research works are devoted to the issues of histogenesis and embryogenesis of RMS. The problem of the origin of this type of tumor was studied in detail by Yu. B. Bakhtin and I. N. Shvemberg. The authors believe that there are three main theories of the origin of rhabdomyosarcoma, which provide a rational explanation for the constant detection in such tumors of not only cells of the myogenic type, which to varying degrees exhibit the ability to undergo cytodifferentiation, but also cells of the connective tissue type, synthesizing collagen and other proteins characteristic of connective tissue, as well as cells that have a pronounced morphological similarity to embryonic mesenchyme cells.
According to Yu. B. Bakhtin and I. N. Shvemberg, from the point of view of the theory of dysontogenetic origin of RMS, the mixed nature of skeletal muscle tumors is due to malignancy of embryonic remains that have retained pluripotency. Essentially, this theory is based on Conheim's well-known ideas about the development of tumors from embryonic remains and dystopias. However, today there are numerous experimental facts, under the pressure of which the theory of dysontogenetic origin of tumors of various organs and tissues is pushed into the background.
These facts include, first of all, the successful induction of tumors in adult animals, the development of which was associated exclusively with the malignancy of dormant embryonic remains. Although at present there is no reason to believe that the majority of RMS is of dysontogenetic origin, for certain types of tumors, which include cellular or syncytial structures characteristic of skeletal muscles, it is quite difficult to deny the possibility of such an origin. This applies primarily to ectopic rhabdomyosarcomas and to areas of RMS found in teratoid tumors.
Speaking about the embryonic origin of ectopic RMS, it is possible, with a certain degree of probability, to name the following embryonic cellular elements as sources of the development of striated muscle fibers found in such tumors:
dystopic cells of myotomes, mesenchymal cells determined in the direction of development of skeletal muscles, omnipotent cells of the germinal rudiment, nephrogenic, and possibly also neuroectodermal cellular elements.
In all these cases, it is assumed that the source of the development of ectopic rhabdomyosarcomas are cells that have separated from participation in normal ontogenesis at one of the stages of embryonic development, but before they reach a mature, differentiated state. It is generally accepted that ectopic RMS arise from split off rudiments of muscle tissue and are dysontogenetic neoplasms - hamartoblastomas of transverse muscle tissue.
RMS in skeletal muscles develop from cellular elements of the muscle tissue itself
- myoblasts, connective tissue cells and mesenchymal cells preserved in the endomysium and adventitia of blood vessels.
The morphological characteristics of rhabdomyosarcomas were first presented by R. S. Horn, N. T. Enterline in 1958 and became the basis for identifying variants of rhabdomyosarcomas in the International Classification of Soft Tissue Tumors in Children (WHO, Geneva, 1969).
As stated above, in the WHO International Classification of Soft Tissue Tumors there are 4 types of RMS:
embryonic (including botryoid), alveolar, pleomorphic and mixed. In recent years, two other types of RMS have been identified in the literature: Ewing-like and undifferentiated mesenchymal sarcoma.
Embryonic rhabdomyosarcoma can occur in areas where there is no striated muscle; it is probably associated with dysembryoplasias and is the most common type of tumor in childhood. Among embryonal rhabdomyosarcomas, myxoid, round cell and tufted tumor variants are distinguished, which depends on the predominance of certain cells and fibers in the analyzed microscopic areas.
Alveolar rhabdomyosarcoma
characterized by the presence of pseudoglandular and pseudoalveolar structures that arise due to the presence of connective tissue layers in the tumor, which are bordered by tumor cells.
Pleomorphic rhabdomyosarcoma has the most variegated appearance:
small, round, oval or spindle-shaped cells in various proportions with giant multinucleate elements.
According to the literature, the following frequency of various RMS variants is observed:
embryonic (including botryoid) - 70%, alveolar - 16%, Ewing-like - 7%, undifferentiated mesenchial sarcoma - 6%, pleomorphic type - 1% of cases.
According to the Research Institute of DOG, various types of RMS have been identified:
embryonic (including botryoid) - 73%, alveolar - 9.3%, mixed - 2%, Ewing-like variant - 1.4%. In 14.3% of children, the tumor type could not be determined. Thus, the most common variant of rhabdomyosarcoma in children is the embryonal variant.
Currently, the role of genetic research in the problem of soft tissue sarcomas is of increasing interest. This concerns primarily the issues of the origin of this type of neoplasm. Thus, S. S. Cooper, M. Straffon (1991) presented data on hereditary predisposition to certain soft tissue tumors, both benign and malignant: lipomas, leiomyomatosis, fibromatosis, rhabdomyosarcoma and a number of other neoplasms. Analysis of cytogenetic data obtained from the study of soft tissue tumors made it possible to identify specific chromosome rearrangements characteristic of certain types of soft tissue tumors.
In addition, close attention was paid to gene amplification and tumor suppressor genes in the development of soft tissue neoplasms.
Histogenetically different soft tissue sarcomas have their own favorite locations. Thus, RMS most often affects the head and neck area and the genitourinary system, while synovial sarcomas and fibrosarcomas are more often localized in the trunk and extremities.
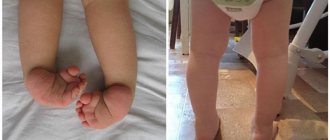
A different frequency of soft tissue rhabdomyosarcomas has been established depending on their location. According to most authors, most often (40 to 50%) RMS are localized in the head and neck region, with the orbit and ENT organs most often affected. Orbital rhabdomyosarcoma accounts for, according to various researchers, from 9 to 37% of cases. The incidence of RMS of the ENT organs varies from 40 to 60% among other malignant neoplasms of this localization. In the area of the trunk and extremities, RMS is observed in 25-37% of cases, and the lower extremities are affected 2.6 times more often than the upper extremities.
The frequency of damage to various parts of the body with rhabdomyosarcoma, according to the Research Institute of DOG, is presented below.
Frequency of lesions of various parts of the body in rhabdomyosarcoma in children
| Localization | Lesion frequency, % |
| Head and neck | 35 |
| Limbs | 25 |
| Genitourinary system | 20 |
| Torso | 10 |
| Other localizations | 10 |
It should be noted that, depending on the location of the primary malignant tumor, two prognostic groups are distinguished:
group 1
- with favorable localization: orbit, paratesticular zone, superficial areas of the head and neck, vagina, vulva, uterus;
Group 2
- with unfavorable localization: trunk, limbs, bladder, prostate, retroperitoneal region, abdominal and thoracic cavities, deep-seated tumors of the head and neck.
Rhabdomyosarcomas are diagnosed more often (80%) in children under 5 years of age, approximately equally often in boys and girls, while synovial sarcomas and fibrosarcomas are more common in children aged 5 to 14 years.
general information
Soft tissue sarcomas are rare diseases in oncology. Soft tissues are understood as all tissues of the body that develop from the embryonic mesenchyme and neuroectoderm: smooth and striated muscles, blood vessels, ligaments and tendon apparatus, cartilage and adipose tissue, as well as peripheral nerves. Sarcomas combine about 30 different morphological variants of tumors and make up only 1% of all malignant neoplasms.
Rhabdomyosarcoma is a malignant neoplasm that develops from the progenitor cells of striated (skeletal) muscle. In pediatric oncology, it is considered one of the most malignant neoplasms with a poor prognosis. Rhabdomyosarcoma accounts for about 4% of all tumors in children, with 60-70% of clinical cases occurring before the age of 10 years. The disease is much less common in adolescents and adults.
The incidence may depend on the histological subtype of rhabdomyosarcoma as follows:
- Embryonic. Patients with embryonal rhabdomyosarcoma are predominantly male (male to female ratio 1.5). The incidence peaks in the 0 to 4 age group at approximately 4 cases per 1 million children, with lower rates among adolescents at approximately 1.5 cases per 1 million adolescents.
- Alveolar. The incidence of alveolar rhabdomyosarcoma does not depend on gender and is constant from 0 to 19 years of age, approximately 1 case per 1 million children and adolescents.
Causes and mechanism of development
The exact causes of rhabdomyosarcoma remain unknown. According to some authors, the leading factor in the development of the disease is hereditary predisposition. Secondary causes of the appearance of malignant neoplasms include:
- influence of radiation;
- exposure to chemical, food and industrial carcinogens;
- congenital and acquired immunodeficiency conditions;
- soft tissue injuries;
- presence of bad habits, passive smoking.
Under the influence of one or more of these factors, cellular mutations occur that are beyond the control of the body's immune system. In the initial stages, rhabdomyosarcoma is a microscopic nodule consisting of atypical (modified) cells. Subsequently, their uncontrolled division occurs, which leads to an increase in malignant neoplasm.
Most histologically confirmed rhabdomyosarcomas have extremely rapid growth and an aggressive course with invasion into surrounding healthy tissue. From a morphological point of view, the problem of malignant neoplasms is the absence of a capsule delimiting the tumor. Although rhabdomyosarcoma has clear contours, the progression of the disease and tumor growth is always accompanied by invasion and infiltration of surrounding healthy tissue. The tumor node grows in several ways: into the thickness of the striated muscles, through the intermuscular spaces, as well as along the fascia (muscle sheaths), nerves or blood vessels.
Rhabdomyosarcoma
Rhabdomyosarcoma is a neoplasm that has a malignant course and originates from skeletal muscle. Against the background of this pathology, uncontrolled growth and division of cells that make up skeletal muscles and are called myocytes occurs.
The reasons for the formation of such a disease are completely unknown today. Nevertheless, oncologists identify several provoking factors, including complicated heredity, high doses of radiation suffered during gestation and the influence of toxic substances.
The basis of the symptoms is the deformation of the affected area, against the background of which signs characteristic of a particular localization of the pathological process will appear. The clinical picture may include nasal congestion, hearing loss, protrusion of the eye and enlarged regional lymph nodes.
Due to the presence of specific symptoms, there are no problems with establishing the correct diagnosis. However, diagnosis should include a physical examination and laboratory and instrumental examinations.
Treatment of the disease involves the implementation of a whole range of therapeutic techniques, including chemotherapy, radiation therapy and surgery. In addition, a donor organ transplant, namely bone marrow, may be needed.
Kinds
In clinical practice, there are three types of rhabdomyosarcomas: embryonal, alveolar and pleomorphic. The embryonal type of tumor is more common than others and is usually diagnosed in infants and children under 3-5 years of age. Typical locations for such rhabdomyosarcoma are the head and neck region, bladder, vagina, prostate gland and testicles.
Alveolar tumor occurs in adolescents and adults. It is usually located in the area of large muscles of the trunk and limbs (usually on the thigh or lower leg). Pleomorphic rhabdomyosarcoma can grow on any part of the body, but most often appears on the lower extremities. This type of malignant neoplasm is diagnosed mainly in adults.
Classifications
There are several classifications of rhabdomyosarcomas. The international TNM classification has the most complete information about the nature and prevalence of the process:
- T - description of the primary tumor
- Tx - insufficient data to assess malignancy
T1 - tumor no more than 5 cm in any dimension; T1a - located superficially, T1b - located deep in the thickness of healthy tissues;
- T2 - rhabdomyosarcoma more than 5 cm in greatest dimension; subgroups T2a and T2b have similar criteria;
- N - regional (located close to the tumor) lymph nodes
- Nx - insufficient laboratory and instrumental data to assess damage to the lymph nodes;
N0 - no metastases in regional lymph nodes;
- N1 - there are signs of metastasis to one or more lymph nodes;
- M - distant metastases to other organ systems
- Mx - no data on distant metastasis;
M0 - no distant metastatic process;
- M1—distant metastases detected.
Another classification of rhabdomyosarcomas is clinical. It takes into account the extent of the malignancy and the possibility of surgical treatment. Clinical classification divides all patients with rhabdomyosarcomas into 4 groups:
- Group 1 - the malignant process is completely localized, there are no regional or distant metastases, the tumor is amenable to surgical excision;
- Group 2 - rhabdomyosarcoma has localized growth, but cannot be completely removed according to the results of histological examination (there are atypical cells along the edge of tumor excision), there are metastases in regional lymph nodes;
- Group 3 - complete removal of the tumor is impossible, which is determined by the naked eye (for example, ingrowth into large vessels and nerves), there are no signs of distant metastasis;
- Group 4 - surgical treatment is impossible or will not bring a positive result, there are distant metastases.
Depending on the degree of differentiation and maturity of the cells that make up the tumor, two types of rhabdomyosarcomas are distinguished: highly and poorly differentiated. The first type of tumors, as a rule, has a more favorable prognosis. At the same time, rhabdomyosarcomas with a low degree of differentiation are characterized by an extremely aggressive course and quickly metastasize to regional lymph nodes and other organs.
The assessment of malignancy is carried out by a pathologist during a histological examination according to the special Grade classification. It reflects the degree of visual similarity of the tumor to normal striated muscles. The less the cells of a malignant neoplasm are similar to muscle cells, the lower the differentiation and the higher the malignancy of rhabdomyosarcoma. Grade 1 tumors are considered highly differentiated; all others are classified as sarcomas of medium or low maturity.
Of great clinical importance is the division of rhabdomyosarcomas of the head and neck region into two groups: parameningeal and non-parameningeal. The first type includes malignant tumors localized in the nasal cavity, ear or nasopharynx, as well as in the pterygopalatine or infratemporal fossa, the second type includes all others.
Which doctor should I contact?
The disease is treated by an oncologist. However, its first signs are most often detected by a pediatrician. Depending on the location of the tumor, consultations with an ophthalmologist, ENT doctor, pulmonologist, gastroenterologist, neurologist or urologist are prescribed.
Embryonic rhabdomyosarcoma is one of the most dangerous cancers and is often detected in late stages. To treat such neoplasms, complex treatment is carried out, which should begin as early as possible.
Rating: (votes - 1 , average: 5.00 out of 5)
Symptoms
Clinical manifestations of rhabdomyosarcoma depend on several factors:
- tumor localization;
- size and prevalence of processes;
- patient's age.
In the early stages, the disease does not have any symptoms. Characteristic clinical symptoms appear as rhabdomyosarcoma grows, when it can be detected with the naked eye or it begins to compress the surrounding healthy tissue.
The most common reason for seeking medical help is the appearance of a single painless tumor on the body. Typical localization of rhabdomyosarcoma is the face and neck, the anterior and posterior surface of the thigh, the calf muscle area, the upper and middle third of the arm. In approximately 2/3 of patients, the presence of a palpable tumor is the first and only symptom of the disease.
With careful palpation, you can identify the following signs of a malignant neoplasm:
- round or oval shape;
- elastic or densely elastic consistency;
- Smooth surface;
- clear contours;
- immovability;
- painlessness on palpation (palpation).
With superficial localization of rhabdomyosarcoma in the muscles of the limb, it may slightly shift under the fingers in the transverse direction in both directions. When the muscles contract, the tumor becomes completely motionless. In the case of an intermuscular location of the malignant neoplasm, palpation is possible only with complete relaxation of the limb; with muscle contraction, rhabdomyosarcoma loses its outline.
In the initial stages, the skin over the affected area is not changed. Subsequently, a local increase in temperature, slight redness and swelling appears. As rhabdomyosarcoma grows, the skin over it becomes thinner, acquires a waxy sheen and has a pronounced vascular pattern. In later stages, it may acquire a purplish-bluish tint. When a malignant tumor grows into the subcutaneous fat and skin or the tumor disintegrates, ulceration of the skin, bleeding, and non-healing of wounds may occur.
As the disease progresses, some patients also complain of pain or dysfunction of the affected part of the body. This is due to compression or ingrowth into the neurovascular bundles, germination into the joint capsule or periosteum.
If the tumor is localized in the area of the ENT organs, nonspecific symptoms may appear in the form of congestion in the ears, difficulty in nasal breathing, mild nosebleeds, and others. Often, damage to the ENT organs is regarded as an infectious or allergic disease, which leads to late detection of the malignant process.
In the later stages of rhabdomyosarcoma, symptoms of cancer intoxication appear. They are caused by tumor growth, metastasis to organs and the release of its decay products into the blood. Clinical manifestations of cancer intoxication include weight loss to the point of exhaustion, weakness, loss of appetite, gastrointestinal dysfunction, and anemia.
Symptoms and signs of rhabdomyosarcoma in childhood
The most important manifestation of rhabdomyosarcoma is swelling in one place or another. There may be no other symptoms at first. They arise later, when the neoplasm moves from soft tissues to nearby structures. For example, when the gastrointestinal or hepatobiliary tract is involved in the oncological process, obstructive jaundice and an enlargement of the abdominal cavity are observed.
Read here: What causes pancreatic cancer in men?
When nerve lines and blood vessels are affected, pain occurs. They can have varying intensity, intensify after physical activity or at rest. Growth into the bone is accompanied by an increase in pain and impaired joint mobility. In advanced cases, a fracture may occur.
Rhabdomyosarcoma of the bladder in children manifests itself in the form of pain in the lower abdomen and pain when urinating. There may be blood in the urine. When the urinary tract is blocked, acute urinary retention develops. Other serious complications include urosepsis and cachexia.
Parameningeal lesions can affect the brain and cause various neurological symptoms as well as intracranial pressure. Facial nerve paresis is not uncommon.
Rhabdomyosarcoma of the scrotum in boys is similar to a hernia or varicocele. Vaginal swelling in girls may be indicated by discharge with an unpleasant odor, as well as pain and itching. The tumor grows quickly and fills the vagina. When stressed, the formation falls out. It looks like a bunch of brown or pink color.
When a tumor grows into the skin, a bleeding ulcer opens.
Other local symptoms of rhabdomyosarcoma in children include:
- hearing impairment, discharge from the ear canal (with damage to the middle ear);
- in the area of the nose and sinuses, rhabdomyosarcoma leads to breathing problems and nosebleeds;
- neck swelling can affect your voice and make it difficult to swallow food;
- if the eye is involved in the process, then there will be a noticeable increase and protrusion of the orbit, as well as deterioration of vision;
- testicular enlargement;
- constipation, diarrhea, nausea, vomiting are signs of gastrointestinal damage.
Common symptoms of rhabdomyosarcoma in children are: poor appetite, weight loss, fatigue, moodiness, poor sleep.
If the following symptoms occur, immediate treatment should be started:
- coma or convulsions (administration of anticonvulsants is indicated);
- severe dehydration or shock (infusion indicated);
- difficulty breathing (oxygen therapy);
- central cyanosis.
The following manifestations of rhabdomyosarcoma in childhood are also considered potentially dangerous:
- severe exhaustion;
- obvious pallor of the palms;
- half-asleep state;
- respiratory distress.
A child's age less than 2 months increases the risk of death!
Diagnostics
The first stage of the examination is the collection of complaints, medical history and physical examination of the patient. Data on how long ago the tumor was discovered by the patient and its growth rate are of important clinical importance. If the growth rate of rhabdomyosarcoma has increased, predisposing factors (trauma, puncture or biopsy of the tumor) must be clarified. After the interview, the doctor feels the tumor and the soft tissue around it, as well as the regional lymph nodes.
The following instrumental diagnostic methods are used to visualize the tumor:
- radiography - targeted “soft” radiographs allow you to determine the shadow of the tumor, homogeneity (homogeneity) and the nature of the contours of the tumor;
- Ultrasound examination of soft tissues is a simple and accessible method for identifying both the primary lesion and its nature, as well as metastatic lesions of the lymph nodes;
- X-ray computed tomography - the method makes it possible to obtain a clear image of not only a malignant neoplasm, but also to evaluate its relationship with surrounding tissues;
- Magnetic resonance imaging - the study helps to obtain the clearest picture of soft tissues and neurovascular bundles in the affected area.
Histological confirmation of the diagnosis is mandatory. For this, various methods of collecting material can be used: tumor puncture for cytological examination, targeted trephine biopsy under ultrasound control, knife biopsy. The simplest puncture is the least informative among them, because The cytological method does not in all cases make it possible to determine the malignant nature and degree of differentiation of the tumor formation.
To identify distant metastases, ultrasound diagnostic methods (abdominal and retroperitoneal organs, lymph nodes), radiography (chest organs, skeletal bones) and tomography (thoracic and abdominal organs, skeletal system) are used.
Treatment
The choice of therapeutic tactics is carried out individually. This takes into account many factors, primarily the patient’s age, histological type, location and size of the malignant neoplasm, as well as the stage of the oncological process.
The following methods are used to treat rhabdomyosarcomas:
- surgical - excision of a malignant neoplasm;
- chemotherapy - courses of drugs that have a cytostatic effect;
- radiation therapy - irradiation of both the tumor itself and foci of metastatic lesions;
- combined - the use of several methods of therapy.
Despite the constant improvement of radiation and chemotherapy techniques, surgery plays a leading role in treatment tactics. The only reason for refusing surgery is the detection of rhabdomyosarcoma in an advanced stage, when there are distant metastases. However, even in this case, palliative (auxiliary) operations can be performed, for example, for large tumors that significantly interfere with the patient’s daily activities.
Surgical interventions are also not used for rhabdomyosarcomas of parameningeal localization due to the high risk of damage to vital anatomical structures. In all other cases, surgery is the main method of treatment.
The goal of surgery is wide excision of the malignancy or “en bloc” resection. A prerequisite is compliance with the principle of sheathing - a tumor growing within the muscular-fascial bed must be removed along with the muscle and fascia covering it. Where, due to anatomical features, this principle cannot be observed, a deviation of at least 5 cm from the edge of the tumor is considered optimal. This is due to the fact that at a short distance from the main focus in supposedly “healthy tissues” there may be microscopic nodules of malignant cells. If they are not removed, the risk of relapse will increase significantly.
Chemotherapy can act as an independent method of treatment or be part of complex therapy. In combination with radiation therapy, it is used in children and adults with parameningeal rhabdomyosarcomas.
Neoadjuvant (preoperative) chemotherapy is given to patients with large rhabdomyosarcomas to shrink their size and reduce the risk of postoperative recurrence.
Adjuvant (postoperative) chemotherapy is not mandatory and is not given to patients with locally advanced disease and a poor prognosis after surgery. It is used to stabilize the oncological process and reduce the size of tumors that cannot be completely excised. The possibility of adjuvant chemotherapy is usually considered in young patients with large superficial or deep tumors that, according to histological examination, have a high degree of malignancy.
Causes of the disease
The causes of rhabdomyosarcoma in children are not precisely known.
Presumably the development of the disease is influenced by:
- radioactive exposure. Oncology can occur at the site where radiation therapy was previously performed. There is also a risk of developing cancer in people exposed to radiation due to poor environmental conditions;
- exposure to chemical carcinogens;
- genetic disorders in DNA and genetic diseases (Li-Fraumeni syndrome, neurofibromatosis, Costello syndrome, Beckwith-Wiedemann syndrome, and Nuan);
- bad heredity;
- various mechanical injuries;
- infections.
Most often, the causes of rhabdomyosarcoma in children are associated with genetic abnormalities. They can be the result of abnormal development in the prenatal period, therefore, in order to protect the child, the mother needs to be careful about her health during pregnancy.
Complications and prognosis
The prognosis of the disease largely depends on the type and size of rhabdomyosarcoma, the timeliness of seeking medical help and the amount of treatment. A malignant neoplasm is characterized by aggressive destructive growth, as well as rapid regional and distant metastasis through lymphatic or blood vessels.
With early detection of the disease and comprehensive treatment, the prognosis is favorable - in 75-80% of patients it is possible to achieve a complete cure or five-year remission. When rhabdomyosarcoma is large, it is often necessary to resort to severe disabling operations. However, even after complex treatment, the risk of disease relapse remains high. If distant metastases are detected, the prognosis is poor. In such cases, symptomatic treatment is carried out aimed at improving the quality of life.
In the European clinic, experienced specialists treat such a serious disease as rhabdomyosarcoma. Thanks to the powerful material and technical base of the clinic and the presence of qualified personnel, we manage to achieve good results even in the most difficult cases.
Book a consultation 24 hours a day
+7+7+78